1. Structure
In nature, lipases (EC 3.1.1.3) or triacylglycerol acyl hydrolases mainly catalyze the total or partial hydrolysis of ester bonds in triglycerides, usually in the aggregate state and at the level of such surfaces, however, other substrates on which they can act in vivo are not ruled out, such as cholesterol esters, phospholipids, lysophospholipids and ceramides, as has been observed for some lipases
[1][2]. Microbial lipases are generally globular enzymes with different sizes: an example of a low molecular weight lipase is that of Bacillus subtilis with 19 kDa
[3], while at the other extreme is lipase A from Serratia marcescens with 64.9 kDa
[4] (
Figure 1). These enzymes may present structural metal cations such as Ca
2+ or Zn
2+; the first related to activity and stability, while the case Zn
2+, with thermostability and a tendency to generate aggregates occurs with the lipases of Geobacillus stearothermophilus or Geobacillus thermocatenulathus
[5][6][7].
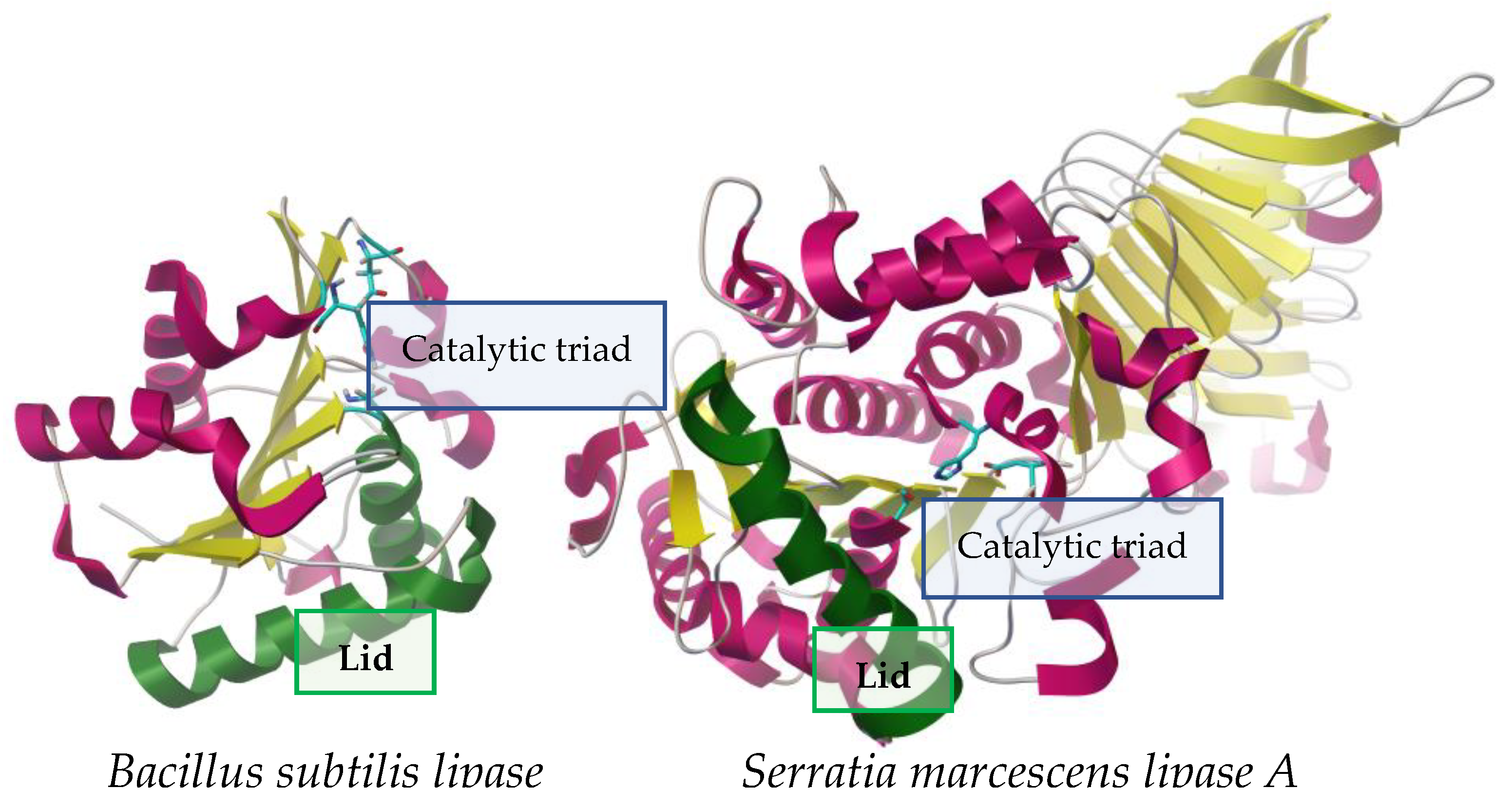
Figure 1. Contrast between two lipases of different sizes from Bacillus subtilis (19 kDa, left)
[3] and Serratia marcescens (64.9 kDa, right)
[4], showing structural similarities such as: lid (represented in green), active center (represented by sticks), α-helices (pink) and β-lamina (yellow). PDB structures 5cri (Bacillus subtilis) and 2aqua (Serratia marcescens), visualized with AutoDockTools 4.2.
Regardless of their origin, lipases are characterized as serine hydrolases in which the catalytic Ser is found within a consensus sequence (Gly-X1-Ser-X2-Gly); this sequence may have variants that, together with the biological origin of the enzyme, will define its classification within the eight proposed families
[8]. The catalytic Ser is part of a set of three amino acids together with His and Asp (or Glu) residues, called the catalytic triad. In the primary structure, this Ser is located closer to the N-terminus, while His is generally located closer to the C-terminus
[9][10][11][12][13][14]. In terms of the secondary structure, the consensus sequence together with the catalytic triad are contained within the α/β hydrolase fold: it consists of a central beta sheet composed of eight parallel beta strands (1 to 8) connected by up to six alpha helices (A–F). In general, in this fold, the Ser is located at the C-terminal end of the β5 strand (within the consensus pentapeptide) characterized by the beta-turn-alpha(αC) motif referred to as the “nucleophilic elbow”. Near this elbow are found the other components of the triad: Asp (or Glu) between αE and β8, which is part of a long independent loop, while His is usually located between αF and β8 as part of a long loop
[10][11][12][13][14]. The catalytic Ser and amido groups C
α (e.g.
, from Gln and Thr) and OH (from Thr or Tyr) will conform the “oxyanion hole”; this assembly in turn constitutes the deepest end of the predominantly hydrophobic cavity where the oxyanion and the acyl acceptor will join
[15][16].
Sometimes the catalytic cavity will be more or less exposed to the solvent depending on the presence and state of a self-regulatory mobile domain called the lid (or flap)
[17]. The lid has an amphipathic and flexible nature and is composed of one or more α-helices and different loops
[10][11][12][13][14][17]. According to evidence from X-ray structure and molecular dynamics, in the case of lipase from Pseudomonas sp. MIS38, the ability to move this lid would be related to the presence of structural metal ions (Ca
2+ or Zn
2+) that function as hinges or attachment points of the lid depending on the lipase
[13][18]. Thus, the lipases that contain lid will present a conformational equilibrium between an open or closed form, depending on the state in which it is found. This has been supported by the large number of lipases such as those from Thermomyces lanuginosus (TLL), Candida (or Pseudozyma) antarctica (CAL B), Geobacillus thermocatenulatus (BTL2), among others, which have been crystallized in different conformations and where it has been shown displacements and reorganizations in the secondary structure of the lid between one state and another: open conformations have not only been observed in lipases co-crystallized with substrates, detergents or inhibitors, but also when they are forming dimers, in which case the lids would mutually constitute part of the binding interface
[10][11][12][13][14][19]. In addition to this evidence, there are other studies with lipases in solution, which through techniques such as: Single Particle Tracking (SPT,
Section 2.2.2), Site-Directed Spin Labeling (SDSL), Site-Directed Fluorescence Labeling (SDFL) and Electron Paramagnetic Resonance Spectroscopy (EPR), have shown that lid-containing lipases present different conformational states depending on the presence of substrates, detergents and, in general, substances that, upon aggregation, assemble hydrophobic interfaces with water
[20][21][22]; this behavior has also been evidenced in silico through molecular dynamics studies
[23][24][25].
2. Mechanism of Action: Interfacial Activation and Catalysis
2.1. Interfacial Activation
As is known, the natural substrates of lipases are mainly triglycerides of fatty acids (TG) characterized by their low solubility in water where they tend to form aggregates. On the other hand, microbial lipases are usually produced as globular enzymes that tend to be soluble in the cellular or extracellular environment of an aqueous nature. This means that, evolutionarily, lipases must have acquired characteristics that allowed them to access these insoluble substrates from the aqueous medium, which necessarily involves a transition to a more hydrophobic conformation through structural changes that do not result in their inactivation or denaturation, but instead lead to another more active folded state: this explains, in part, why some lipases are stable even in the presence of non-aqueous solvents unlike other enzymes such as aldolases (See Molecular Origins of the Catalytic Promiscuity of Lipases)
[26].
The above implies that in aqueous media or in the absence of substrates (and their analogs), the majority population of lipase molecules (with lid or flap) shows a mainly closed conformation where the lid interacts through hydrophobic side chains with the outermost part of the catalytic cavity
[13][17]. In contrast, in the presence of aggregated substrates, lipase experiences an increase in the proportion of its hydrophobic area characterized by a new conformation that constitutes the dominant population of lipase
[17][20][23][27].
The presence of a TG-water interface and an adequate orientation of the lipase
[28] towards the surface of TGs would shift the conformational equilibrium to a form in which the lid domain constitutes part of the interaction surface with the substrate surface added, leaving the catalytic cavity accessible, resulting in an increase in activity. The main thermodynamic engine of the phenomenon described is the so-called “hydrophobic effect” (
Figure 2)
[29]: the displacement of clathrate water molecules from the hydrophobic surface of the adsorbent (here the one made up of aggregates of TG molecules) generated by the adsorbate (herein, the enzyme) produces an increase in the entropy of the system. This not only generates an increase in the detected catalytic activity or “interphase activation” but may also imply the stabilization of the adsorbed lipase as a consequence of the reduction in the conformational space available for the unfolded state
[30]: this type of phenomenon of activation/stabilization would also occur in the case of lipases immobilized on macroporous solid supports of a hydrophobic nature, as will be seen later.
Figure 2. Scheme of open-closed conformational equilibrium of lipases (i.e.
, BTL2, PDB 2w22). Upper panel (equilibrium in water): closed form (
upper left) and the less populated open form (
upper right). Yellow and purple spheres represents lipase structural ions. Blue spheres represent clathrate water molecules and “T” for triglycerides, both at the interphase. Lower panel: after lipase adsorption, some clathrate water molecules get desorbed and disordered as bulk water. Some individual “T” (substrate) molecules can now diffuse towards the active center of the more stable open lipase form (
lower right), starting catalysis
[28].
2.2. Catalysis of Lipases Functioning as Serine Hydrolases
Once the lipase is adsorbed and its open conformation is stabilized, the transport of substrate molecules from the interface to its catalytic cavity will be favored; this has been evidenced by molecular dynamics studies where the M37 lipase in the open state acquires an angled position in front of the hydrophobic aggregated substrate surface and from there a molecule of the substrate is “pulled” until it is located in the active center, finally forming the Michaelis complex
[25]. Although it is not explicitly indicated why the substrate molecule is “pulled”, it is understood that it is positioned in such a way that its reactive groups, of a more polar nature, are attracted towards the area of the catalytic cavity more polar, in other words, where the catalytic Ser and the oxyanion hole are.
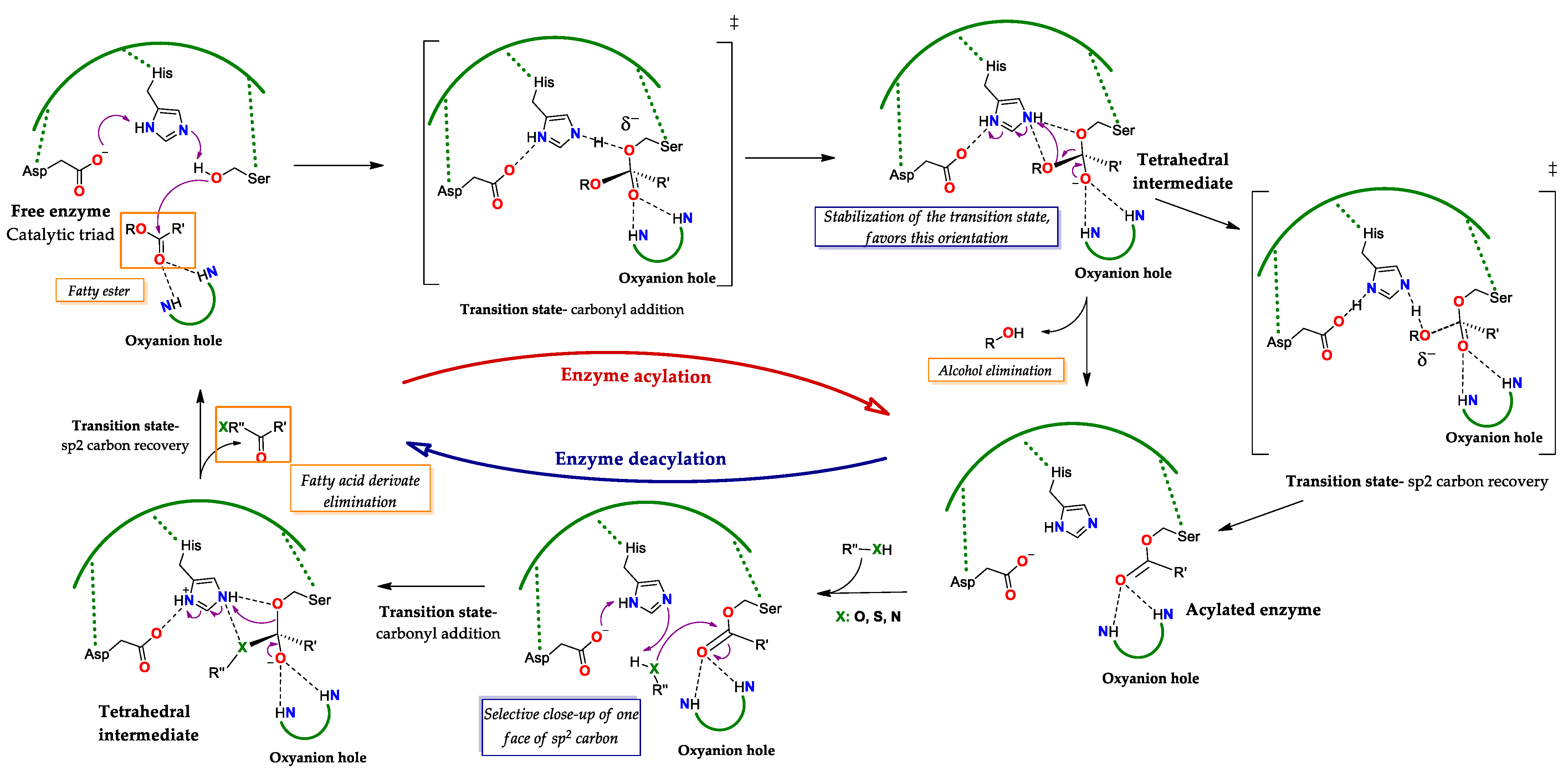
Regarding the Michaelis complex, QM/MM studies have shown that the formation of E-S
1 occurs with the “acyl donor” substrate and involves polar interactions between its O carbonyl with three H bonds donor groups in the oxyanion hole: two –NH residues from amino acids of the peptide chain, such as Gln and Thr, and one with the β-OH group of the side chain of the latter. As a consequence, the C carbonyl remains close to the O(β)H group of the catalytic Ser (
Figure 3): It is important to point out that the interactions between the oxyanion hole as H bond donor and as carbonyl O acceptor will be maintained in the different transition states and intermediates
[31][32][33]. These studies have shown that lipases, such as CAL B (or PAL B), employ a concerted mechanism; here, the transition state of reaction of the O(β) with the C carbonyl of S
1 proceeds as H forms a bond of hydrogen with the imidazole N(ε) of the His of the triad, which supposes the shortening of the bond between the N(δ)H and the carboxylate (γ) of the Asp of the triad
[31]. As a result, the transition state the C carbonyl of S1 becomes polarized, adopting a sp
3 configuration and the O carbonyl acquires a negative charge. Thus, the tetrahedral intermediate results from the formation of the covalent bond between the O(β) of Ser with the now sp
3 carbon (formerly sp
2 carbonyl) of the substrate, the acquisition of negative charge of the carbonyl O (oxyanion) and the protonation of the N(ε) of His and of the carboxylate γ of Asp within the triad (
Figure 3). This intermediate undergoes a charge rearrangement that involves the formation of a second transition state, which leads to the exit of the alkoxy residue from the substrate to which H is transferred from the N(ε) group of His, and, in the formation of an ester-type bond between Ser and the acyl moiety of the substrate, an intermediate known as “acylated enzyme”. The arrival of the acyl acceptor substrate “S
2” of a nucleophilic nature (normally water), implies the formation of a third transition state, in which the S
2 molecule forms interactions as a donor of an H bond with the basic N(ε) of His and as a donor of an electron pair of its O atom with the carbonyl C of S
1 (in the acylated enzyme), causing the loss of its sp
2 character (
Figure 3). This last interaction leads to the formation of a covalent bond between the O of S
2 with the C carbonyl of S
1, forming a second tetrahedral oxyanion-type intermediate and the protonation of N(ε). The weakening of the bond between the O(β) of Ser and the C carbonyl of S
1 occurs at the same time as an interaction with the H of N(ε) and a transition from sp
3 to sp
2 in the C carbonyl is established, which generates the last transition state. Finally, the formation of the acylated product is achieved by breaking the O(β) bond with the C carbonyl while the protonation of the catalytic Ser occurs, reestablishing the initial state of the enzymatic triad and the release of the product (
Figure 3). These QM/MM studies are consistent with the Ping-Pong Bi-Bi mechanism used to kinetically model reactions catalyzed by free or even adsorbed (immobilized) lipases on hydrophobic substrate surfaces, reactions including not only hydrolysis but also alcoholysis, acidolysis and transesterification
[34].
Figure 3. General catalytic cycle of lipases. The reaction starts by the nucleophilic attack of the catalytic Ser to the substrate, passing through a transition state (‡) of addition to the carbonyl to the generation of the selective tetrahedral intermediate (here, the R stereoisomer) where the negative charge density is concentrated in the O(β) of the Ser, here the preference towards the stereoisomer R is explained by the fact that the most stable transition state (‡) is the one where this orientation is favored. Next, the sp
2 carbon is recovered, through a transition state (‡) here the charge density is focused on the O of the alkoxy group to be eliminated, in order to obtain the acylated enzyme. Subsequently, in the presence of a nucleophilic molecule type RXH, where X can be O, S or N, a tetrahedral intermediate is regenerated, by the addition transition state (‡) to one face of the sp
2 carbon of the carbonyl. Finally, the sp
2 carbon is recovered, producing a fatty acid derivative and regenerating the free active site. Adapted from
[31][32][33].
Interestingly, something not well studied is that the consequences of the catalysis phenomenon described above, which focuses within the catalytic cavity, also seems to have a reflection outside of it: using the Single Particle Tracking (SPT) assay, it was determined that in the extent that lipases (e.g.
, TLL) catalyze the hydrolysis of aggregated triglycerides, the products of the reaction (alcohols and acids fatty) would dynamically affect the state of the adsorbed lipase: these products would function as “lipase repellent coatings”, so that the enzyme diffuses towards more hydrophobic surfaces, a kind of “chemotaxis” guiding the lipase to surfaces where the concentration of the unprocessed substrate is higher, thus favoring a higher catalytic efficiency
[22]. These types of studies open an opportunity to study thermodynamic and kinetic aspects less explored in the catalysis of lipases, such as, for example, how the composition of the interface on which the enzyme is adsorbed, or lid modifications, affect the activity or selectivity of the lipase. This would have applications in the production of APIs that facilitate their production in the presence of interfaces, for example, as occurs in the kinetic resolution of esters containing azaspirononanes or spiro-β-lactams by the lipase of
Arthrobacter sp.
[35], respectively. These compounds were resolved in a biphasic system of diisopyopyl ether/phosphate buffer and are related to homoharringtonine, a molecule approved for the treatment of chronic myeloid leukemia and in the production of antibiotics
[35][36].
2.3. Selectivity of Lipases
Lipases can catalyze reactions involving natural lipids that, in addition to TGs, can include the group of phospholipids: some of these are bioactive (phosphatidic acid or lysophosphatidylcholine) and others, such as lecithins, are used in the pharmaceutical industry as emulsifiers or for the release of active principles when they are part of liposomes, such as those present in the first nanodrug approved by the FDA, the anticancer Doxil
® [37][38]. Given their structural similarity, these phospholipids can be obtained naturally or synthetically from TGs as sources of fatty acids, some of which are of interest in human health, such as PUFAs (polyunsaturated fatty acids)
[39]. Other derivatives have also shown potential as prodrugs to mitigate the side effects of mefenamic acid, an anti-inflammatory
[40]. Due to the great variety of natural TGs and their abundance in biomass, it has been important to establish connections between their structure and the observations obtained during their enzymatic transformation in order to obtain derivatives of commercial interest. This has enabled a greater understanding of the structural origins of the selectivity of lipases and extrapolate some of their implications on synthetic substrates, as shown below:
Chain Length
It has been observed how the lid and its dynamics influence substrate selectivity in lipases
[17], also conditioning the adsorption of TLL on the hydrophobic surface of substrates (orientation, activation and stability) and its diffusion on it
[22][24]. It is important to note that not only the catalytic cavity but also the lid can be part of such substrate binding surfaces. The structural model of Pleiss et al. have postulated that the size, polarity, and shape of the lipase catalytic cavity (crevice, funnel, or tunnel) will determine the regio- and enantioselectivity of the lipase
[16]. Through structural comparisons between a group of eight esterases and lipases, they showed that the catalytic cavity of the latter is usually larger, essentially to accommodate comparatively more hydrophobic and larger substrates such as medium and long chain TG. For example, in esterification or alcoholysis reactions, the fact that CAL B (or PAL B) had a cleavage site for the acyl residue (scissile fatty acid binding site) at the height of the carbon chain up to the C13 carbon atom while that the Rhizomucor miehei lipase (RML) presented it up to C18 explained the preference of the latter for longer chain substrates
[16].
Mutant lipases that present blockades in scissile sites of the acyl residue or that are more hydrophilic tend to decrease their preference for long-chain TGs (triolein in relation to short-chain substrates (tributyrin)) in lipases from Rhyzopus sp.
[41]; something similar was observed with variants (Gly237Ala/Leu/Val/Tyr) of CAL A, modified by directed mutagenesis that generate blockages at the entrance of a binding tunnel where part of the long chains of the substrate were accommodated in the native enzyme
[42]. Lately
[43], for this enzyme, the 217–245 helix-loop-helix motif was identified as a key in TG recognition: mutants with a single substitution that conferred discrimination between short versus long TGs, for this enzyme, the 217–245 helix-loop-helix motif was identified as a key in TG recognition: mutants with a single substitution that conferred discrimination between short versus long TGs were identified at positions 183, 235, 240, 244, 338 and 377, while those with a preference for long TGs had substitutions at positions 84, 93, 232 and 359.
Cis/Trans Selectivity
Structural evidence of this type of selectivity has been obtained by studying lipases of known structure in esterification reactions against geometric isomers of 9-octadecenoic acid in cis (oleic) and trans (elaidic) and with cis/trans and positional isomers of conjugated linoleic acid (CLA)
[44][45]: in general, and following the analyzes of Pleiss et al., enzymes with wider binding cavities are better at catalyzing reactions of the cis vs. trans isomer of 9-octadecenoic acid as RML, while those with deeper and narrower cavities (fit better with the trans isomer) such as CAL A show a higher trans/cis selectivity compared to the previous enzyme; in the case of the CLA isomers, no clear trends were observed, since, due to the conformations that such substrates could acquire, not many differences are expected in terms of their shape. In other study
[46], it was observed that the Phe149, Tyr221, Phe222, Ile301, and Leu305 residues were crucial in the generation of binding tunnels to straight or curved acyl residues, favoring selectivity for trans (or saturated) or cis fatty acids, respectively.
Preferences between tri, di- and Mono Glycerides
Within the lipases, there are also differences in the chemoselectivity of the type of glyceride they prefer to react with. More selective lipases against partial glycerides (mono- and diacylglycerol lipases, MDGLs) have a wide interest in applications in the food, nutraceutical and biofuel industries
[47][48]. In Malassezia globosa lipase (SMG1), the selectivity against mono- and di-glycerides would result from the presence of two bulky and hydrophobic residues (Trp, Phe) adjacent to the catalytic site and a long N-terminal hinge region of the lid that can interact as a steric hindrance for the entry of TGs
[49]. A mutant Q282L of SMG1 revealed that Gln in the native enzyme, being less hydrophobic than Leu in the mutated one, would be partly responsible for a less favorable interaction with TGs, explaining the null activity of the native enzyme against the latter
[50].
Taking as reference the Aspergillus oryzae MDGL-like lipase (AOL), also with bulky residues (Trp, Phe), at the entrance of the catalytic cavity, a consensus sequence Phe-X
1-X
2-His was identified for the MDGLs of the RML family. This sequence would form a “bridge-like” structure that would govern the recognition of substrates by preventing the accommodation of the sn-2 chain in the case of a TG
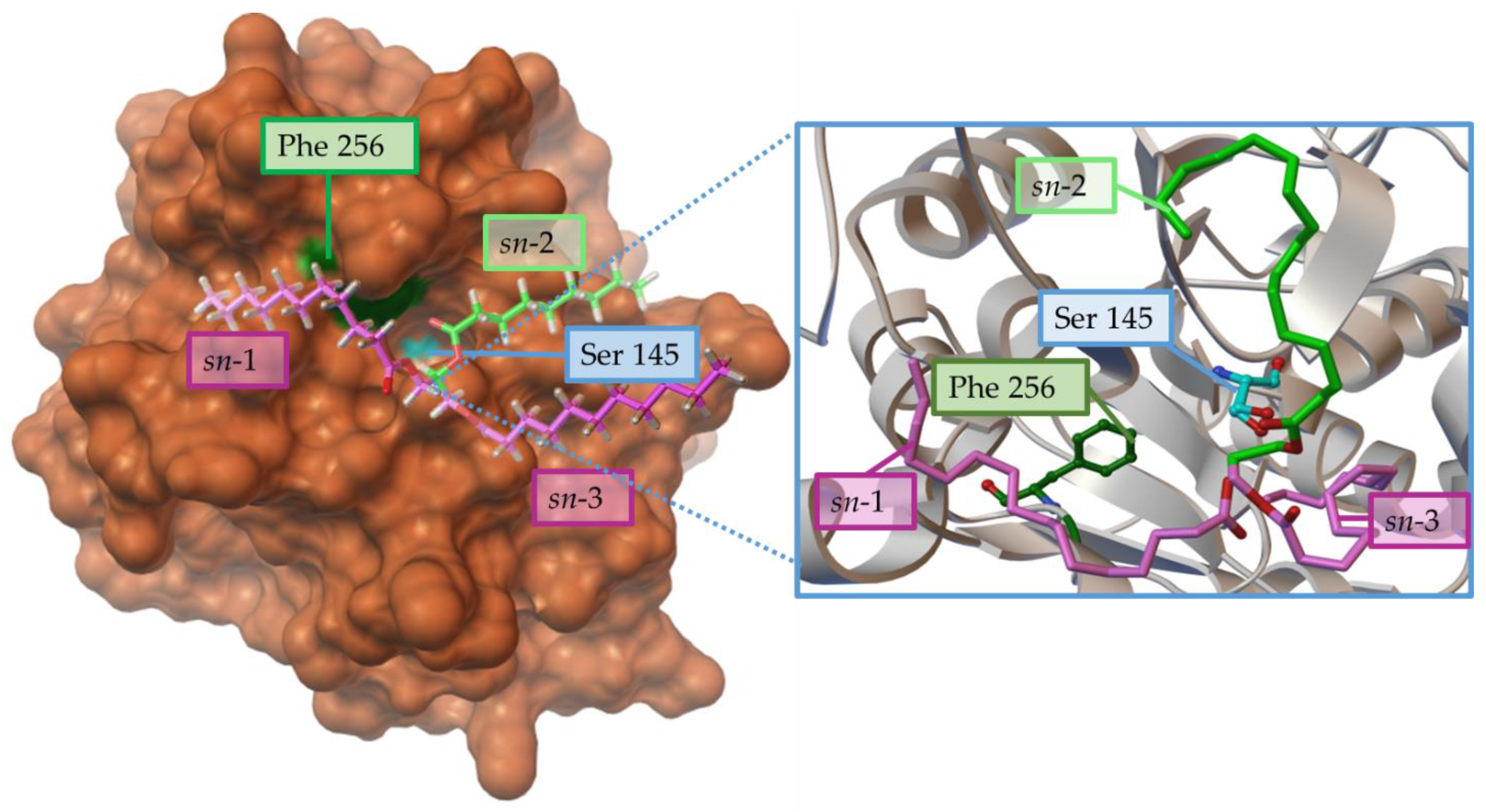
[51]. Something similar has been observed in the recombinant lipase from Penicillium cyclopium (rePcMdl), where the Phe256 residue would prevent an adequate interaction from the sn-1 chain of the TG (
Figure 4).
Figure 4. Steric effects of the residue Phe256 (Green) from Penicillium cyclopium lipase prevent the adequate interaction of sn-1 position (violet) in the substrate triacylglycerol (cyan) with the catalytic triad represented in orange, but this effect about the carboxyl carbon from the sn-2 position to the O(β) of Ser145. Structure from PDB 5ch8, using AutoDockTools 4.0 viewer and adapted from
[51].
Regioselectivity
As indicated, in nature TGs are the natural substrates of lipases. Due to their structure they have three positions (stereospecific numbering system) through which they can be hydrolyzed, defined as sn-1, sn-2 and sn-3. Thus, lipases have been classified by their ability to hydrolyze positions 1 and 3 that are equivalent (sn-1,3 selective), position 2 (sn-2 selective) or nonselective. Lipases able to introduce palmitic acid in sn-2 position are of interest in the infant food industry, given their ability to produce from vegetable sources triglycerides similar to those found in human milk
[52]. The sn-1,3 selectivity shown by lipases as those from Rhizopus oryzae (Lipase F AP-15
®) in hydrolysis or in acylations, has enabled their use in applications such as in the production of phospholipids rich in DHA (docosahexaenoic acid) or in nutraceuticals with potential benefits for human health
[37][53].
In studies with CAL B using kinetic measurements and molecular dynamics studies, an attempt was made to explain the influence of the solvent on its ability to esterify sn-1 or sn-2 positions of glycerol with oleic acid. It was observed that the sn-2 position tended to be favored proportionally to the log P of the solvent. This was due to an increase in electronic and Van der Waals interactions between the enzyme and the sn-2 position and a weakening with the sn-1 one, although, in absolute terms, the latter remained in the most stable position
[54].
According to the mechanism described for serine hydrolases in the case of an esterification with glycerol, the enzyme is expected to form a covalent intermediate where the acyl group is relatively fixed by its interactions with the oxyanion hole and the catalytic Ser, while glycerol, in accordance with its orientation in the pocket where it resides, presents interactions with the nearby environment, especially an H bond as a donor with N(ε) of His that can be given from its –OH groups in position 1 or 2. Taking into account the above, the reason why the sn-1,3-selective lipases are more numerous (RML, TLL, CAL B, etc.) than the sn-2 or non-selective ones such as CAL A or that of Geotrichum candidum
[52][55] could be that the positions that have primary OH groups in glycerol are more abundant, reactive and less hindered (assuming similar acyl lengths), so that acquiring an adequate orientation towards N(ε) of His would be more likely, favoring the sn-1,3 selectivity
[56].
Regarding this type of positional selectivity, it must be taken into account that it can be affected by collateral reactions such as acyl migration present in partial glycerides. These migrations can occur spontaneously (especially at high temperatures) or be catalyzed by lipases at room temperature; the latter has been shown in density functional theory (DFT) studies
[57]. An increase in acyl migration can also occur due to reaction conditions such as the use of polar solvents, high a
w or silica-type immobilization supports, etc.
[58][59]. These aspects must be taken into account in API synthesis processes (or their building blocks) that generally contain adjacent –OH groups, as in the case of the presence of sugars and their derivatives
[60][61] and in other polyols: an example is the case of derivatives of Flurbiprofenate, a powerful nonsteroidal anti-inflammatory drug (NSAIDs), which has been esterified with L-ascorbic acid (a polyhydroxy acid) through the use of different lipases (CRL, PPL and Novozym
® 435) in order to improve its absorption in the brain
[62].
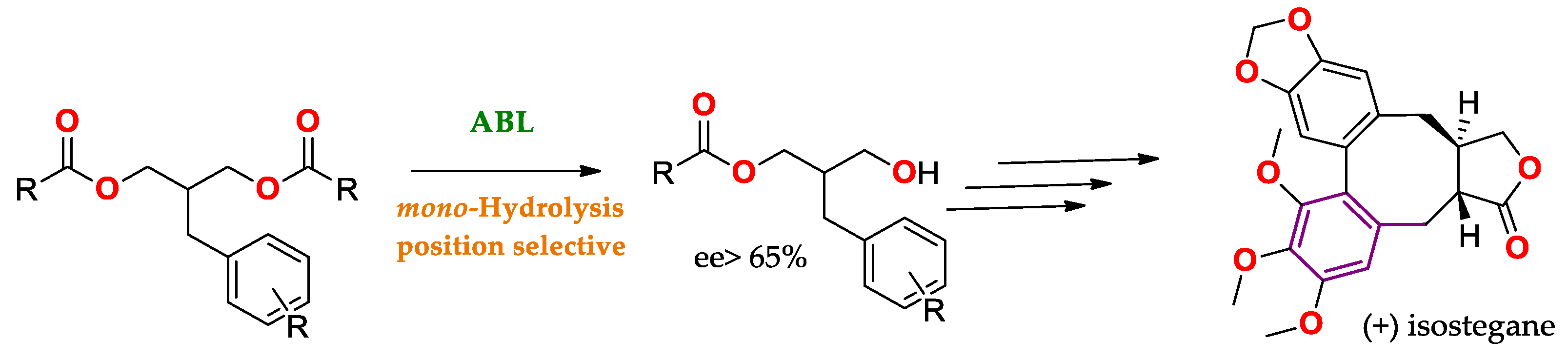
Another example of the use of lipase regioselectivity to obtain APIs is represented in Scheme 1, where mono-hydrolysis is allowed to obtain a precursor of (+) isostegane
[35] from a prochiral substrate (see Enantiomeric/Prochiral Preference (Regioselectivity in Chiral Triglycerides)).
Scheme 1. Mono-hydrolysis using ABL for the production of a precursor of (+) isostegane
[35].
In summary, in processes where regioselectivity is sought, the structural and chemical restrictions of each enzyme, substrates and the particular dynamics of the catalytic phenomenon must be taken into account. This includes the adsorption of the enzyme at the interface constituted by insoluble substrates and the thermodynamic parameters of the reaction. Together, these complexities have made it difficult to establish universal relationships between structure and regioselectivity in lipases with their natural or synthetic substrates
[52], thus constituting a knowledge gap to be solved in the subject.
Enantiomeric/Prochiral Preference (Regioselectivity in Chiral Triglycerides)
Undoubtedly, finding chemical or physical processes that favor the enrichment of products with defined chirality and high purity is a continuous operation in synthesis laboratories and in the pharmaceutical industry. As is known, the guaranteed high purity of chiral active principles for drugs implies greater safety for the consumer. Once the intensification and optimization of the process to obtain those APIs has been achieved (with the consequent reduction in costs and environmental impacts), the sustainability of the pharmaceutical industry will be strengthened
[63][64], which is significant considering that this industry is one of those facing the greatest challenges to comply with the principles of green chemistry
[65].
Due to their advantages, microbial lipases have had a leading role in the solutions to these challenges through the different reactions that they catalyze by favoring reaction paths that lead to processing substrates and obtaining products of defined chirality through strategies such as kinetic resolution
[66][67]. This selectivity has been quantified through parameters such as the
enantiomeric excess percentage (
ee%), which is the difference between the percentage of the major enantiomer and
percentage of the minor enantiomer produced in a reaction mixture
. It is related to the enantiomer ratio
(E), which depends on the relative reaction rate of an enantiomer (in a racemate) to that of the other
, using a biocatalyst under kinetically controlled conditions (e.g.
, first-order or pseudo-first order regime
): this reflects how much the biocatalyst stabilizes the transition state of a reacting enantiomer with regard to the other
[68][69]. In the recent review by Chen et al., an exhaustive description of the structural origins of enantioselectivity in some lipases was made
[70]. Here, some of the general aspects highlighted in that reference, but they will be complemented by approaches not covered there will be summarized.
Structurally, lipases have two pockets in their catalytic cavity: one for the acyl group and one for the alcohol moiety in the case of an ester. The latter is distinguished by having a median and a larger pocket: the median pocket, also known as the “stereoselectivity pocket”, is located deep within the protein (in the case of CAL B), and the largest pocket constitutes the substrate entrance. Thus, a chiral secondary alcohol with substituents of different sizes (e.g.
, methyl vs. propyl or bigger) can be sterically differentiated
[71], explaining why most lipases tend to prefer R-enantiomeric secondary alcohols (Kazlauska’s rule)
[56]. As will be seen, there are other factors that give rise to exceptions or different interpretations to this rule: the “Mirror-Image Packing” rule has broad support based on crystal structures
[72] or the “Key Region(s) Influencing Enantioselectivity”(KRIE), which is based on a review of papers by its authors
[73] about mutant lipases that improved their enantioselectivity not only towards derivatives of chiral alcohols but also towards those of chiral acids. In the case of KRIE, they concluded that some characteristics are key when designing lipases with improved enantioselectivity, such as the distance between the chiral carbon and the carboxyl or alcohol function, as appropriate, and its interaction with lipase regions such as: (i) the acyl-binding pocket, (ii) the hydrophobic cavity, (iii) the hydrophilic cavity and (iv) the oxyanion hole
[73]. However, to generalize the KRIE, greater experimental support is required, something that will only be achieved by testing it with a greater number of lipases, substrates and reaction conditions.
In conclusion, the enzyme and substrate structure are dynamic variables due to their mutual interaction or due to the reaction conditions during the catalytic process: it is not always possible to attribute the observed selectivity nor to design strategies to improve them based on first principles or by taking into account a single factor. Thus, experience in the laboratory supported by an adequate experimental design will continue to be essential, not only as a criterion to refute or not the validity of the proposed structure-function hypotheses, but also as a source of new knowledge, sometimes unexpected. For example, it was observed that when using certain conditions, a lipase is capable of behaving like an enzyme of a different class, as described below.
2.4. Reaction Specificity of Lipases
As the editor of this Special Issue mentions “the narrow specificity, and low promiscuity, or the high costs of enzyme production usually hamper the industrial applications of enzymes for APIs manufacturing”. Indeed, it is very difficult to ensure that the same enzyme maintains its selectivity (e.g.
, high
ee%) and a high turn-over number within the type of reaction it catalyzes regardless of the structural or chemical variants that the substrates may have. This is precisely due to the degree of specialization reached by some enzymes. On the other hand, as in the case with treatments against cancer or bacterial infections, the pharmaceutical industry is continually looking for new APIs, since those that were initially commercially available must be replaced, either because they have triggered therapeutic resistance, or because they must minimize its collateral or secondary effects
[74][75]. One strategy to find new APIs is to generate modifications on a common chemical template to which the biological activity, known as pharmacophore, is associated. These modifications consist of, e.g.
, introducing new substituents, positional changes, chemical functionalities, etc. For this reason, the enzyme that showed optimal properties for obtaining a building block of an API or the API that is intended to be replaced very probably will not be optimal for all the chemical and structural variants of the pharmacophore, which, in turn, will imply finding a new variant of the enzyme, either by prospection and/or by design, which, if not achieved in an adequate time, could entail replacing this biotransformation with a chemosynthetic step: the latter will normally be associated with a less green production process, something undesirable considering the very high E-factors that characterize this industry
[65]. Of course, there is no single strategy to overcome these obstacles and to make finding biocatalytic variants that have, at least in part, the idealized properties for a catalyst needed in the production of a new API possible: computational models, molecular biology tools, enzymatic immobilization and reaction medium engineering, etc., provide tools to shorten the times in which that goal can be reached.
Another ideal situation for an enzyme to be attractive in industrial applications is that it is capable of presenting optimal properties, not only for the same class of reactions but also for others necessary to obtain an API. At the time, it was taken for granted that the catalytic domain of an enzyme could only catalyze some of the reactions of its class, for example, that hydrolases such as lipases could only carry out EC 3 class reactions. However, at least since 2003
[76], it has been shown that lipases such as CAL B are also capable of carrying out reactions previously assigned to the lyase class (EC 4). This property in an enzyme is known as catalytic promiscuity, although it is not the only promiscuity type (
Figure 6). Next, the focus is on the molecular origins of promiscuity in microbial lipases, a topic covered more extensively in the articles of Kazlauzkas
[77], Gupta et al.
[26][78], Wang et al.
[79] and recently for Dewivedee et al.
[80] and Patti et al.
[81], but with an emphasis on the way in which this property can be modulated so that this versatility may favor lipase application in industry.
Figure 6. Schematic reaction coordinate vs. potential energy diagram for lipases, showing the transition state (‡) for each case. Representation of condition promiscuity: lipase can catalyze the hydrolysis of triglycerides (in the scheme, from the center towards the left), and under adequate circumstances, to catalyze reactions in the opposite direction (esterification). In addition, substrate promiscuity is present (
left): they are capable of hydrolyzing other carboxylic acid derivatives such as amides or synthesize them from amines and carboxylic acids, here it can be seen that the difference between transition states (‡) is precisely the nucleophile involved.
[82].
In addition to the three types of promiscuity previously exposed, a fourth type called “product promiscuity” has recently been considered
[83]. This refers to when the same enzyme is able to convert a single substrate into multiple products in reactions that are independent of each other (
Figure 7). In this sense, this means that for each reaction for the production of such products, it is necessary to go through different transition states
[83]. Although the reactivity of the substrate functional groups have an effect on the regiochemics of the reactions, in particular for biocatalyzed reactions, it has been demonstrated that the conformation adopted by the enzyme prior to the formation of the substrate enzyme complex is that which determines the main product of the reaction, according to recent review
[84]. Thus, depending on the stability of possible conformations, the regioselectivity of a reaction can be predicted. Complementary to this, in the same review
[84], it details the importance and positive effect of conformational flexibility in a study on product promiscuity, using the native species of glutathione transferases and mutants with greater flexibility. It is important to recognize that although there are no reports of the use of lipase product promiscuity for the production of APIs, it must be recognized that this property can be exploited in the development of tandem methodologies towards the synthesis of APIs, where from the same substrate two precursors can be obtained that would react to each other to improve the possibility of obtaining the target molecule.
Figure 7. Schematic reaction coordinate vs. potential energy diagram for lipases. Representing an example of a catalytic promiscuity: the two-half panel, showing that the lipase can act in both directions, and product promiscuity: the enzyme as aldolase when faced with a substrate with two or more reaction possibilities, it is showed in each transition state (‡); as is the represented case of an unsaturated α, β aldehyde, the addition of ketone (in the form of enol) can occur in the β-position carbon or carbonyl carbon. One of the two products will be favored depending on which product favors the more stable conformation of the enzyme
[84].
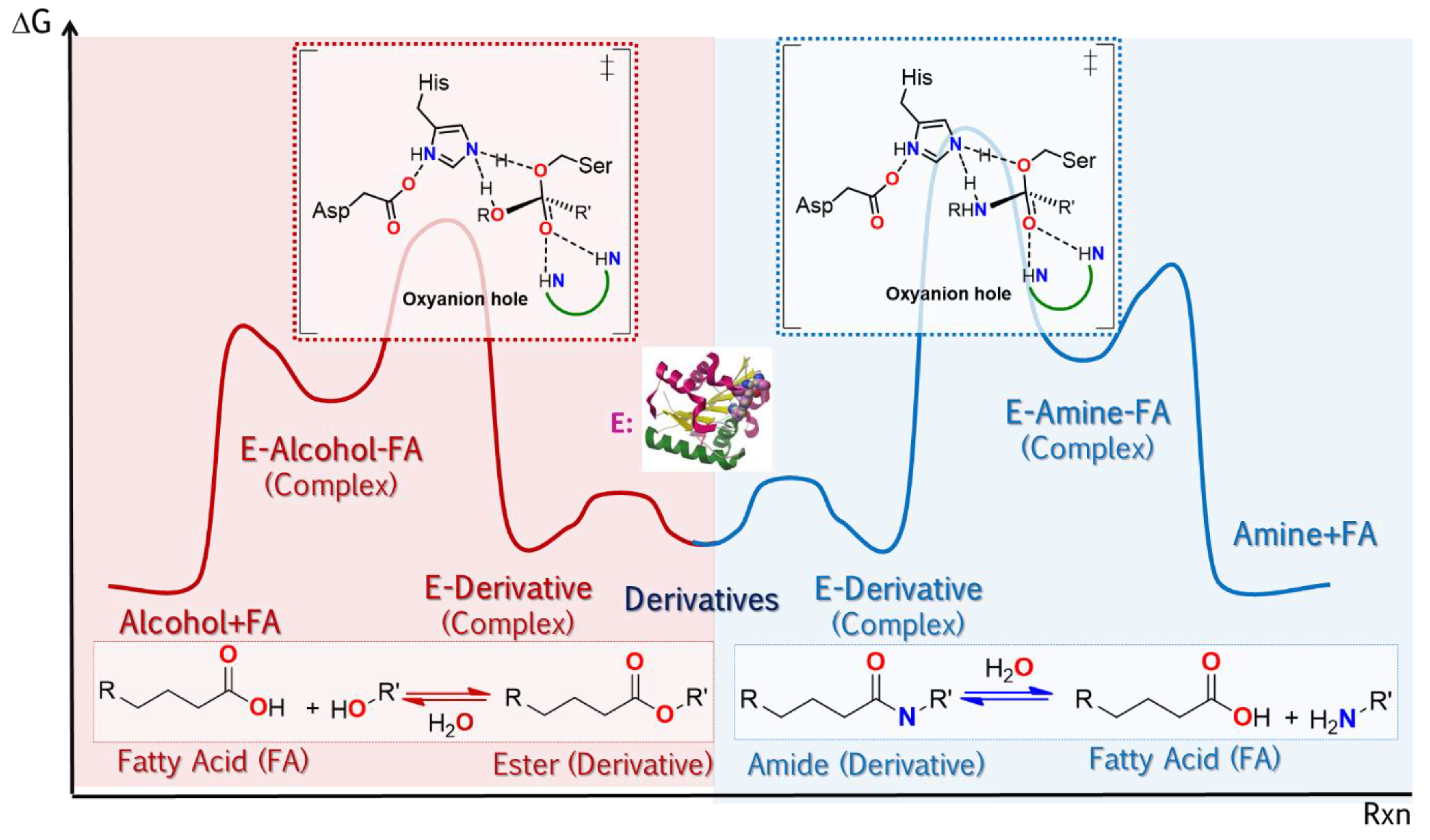
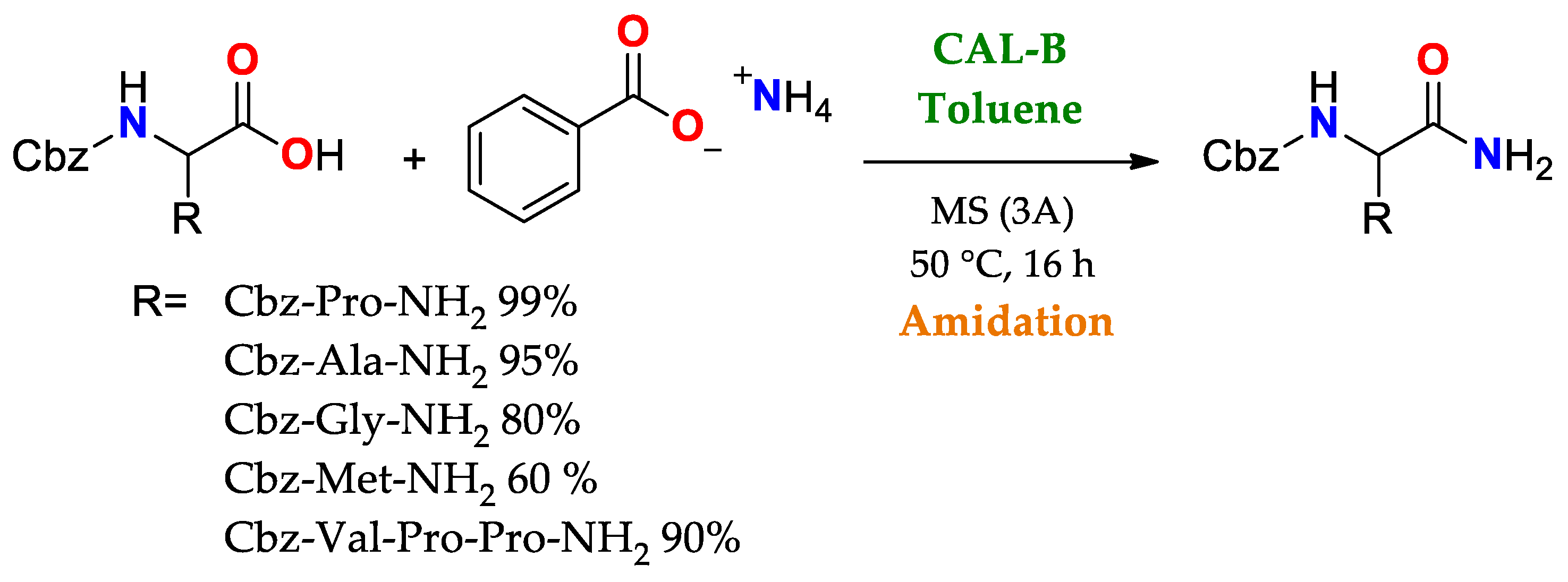
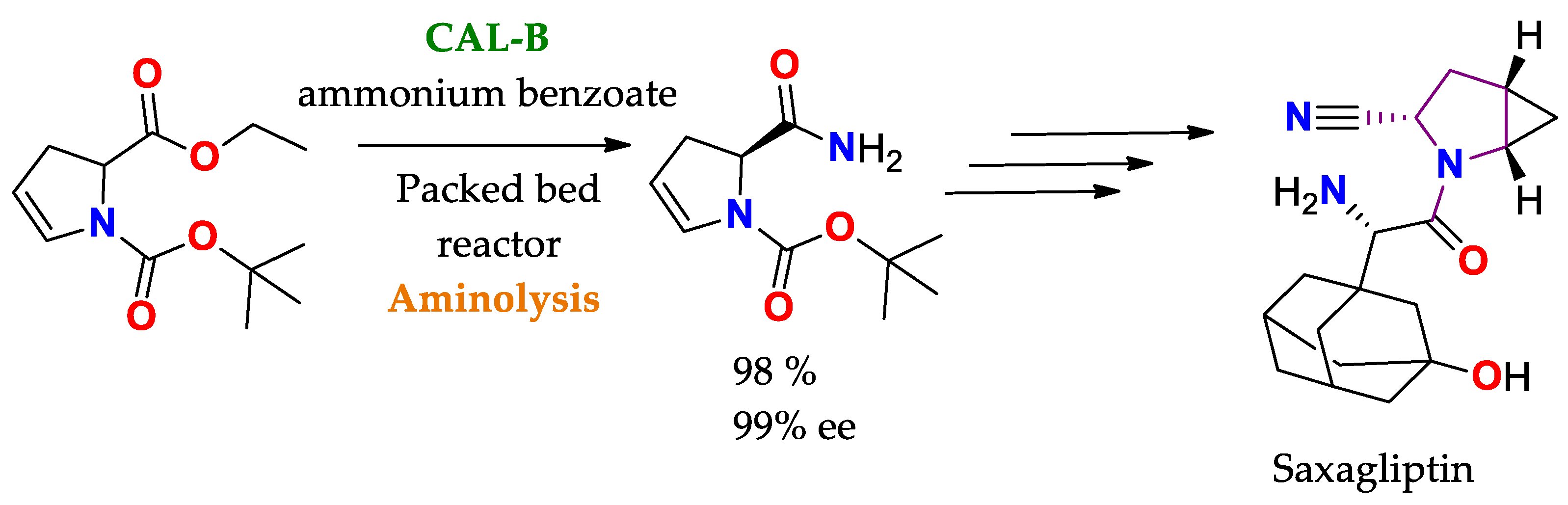
The broad uses of lipases as promiscuous catalysts takes advantage of its substrate promiscuity, such as in selective reactions involving the generation or cleavage of amido bonds. Some cases highlighted here are: i. amidation of amino acids and peptides using CAL-B, Scheme 2, and ii. selective transamidation to obtain a precursor of Saxagliptin (Active ingredient for some diabetes medications), Scheme 3
[85].
Scheme 2. Use of CAL B in the amidation of acid ends of amino acid chains as a key stage in the synthesis of short peptides
[85].
Scheme 3. Selective aminolysis of 1-tert-butyl 2-ethyl 2,3-dihydro-1H-pyrrole-1,2-dicarboxylate to obtain a precursor of Saxagliptin (active drug ingredient for the treatment of diabetes)
[85].
This property has been used to obtain molecules with biological activity. In particular, in the resolution of racemic mixtures to obtain the final product (Scheme 4)
[86] as for APIs resolution (Scheme 5 and Scheme 6)
[86][87].
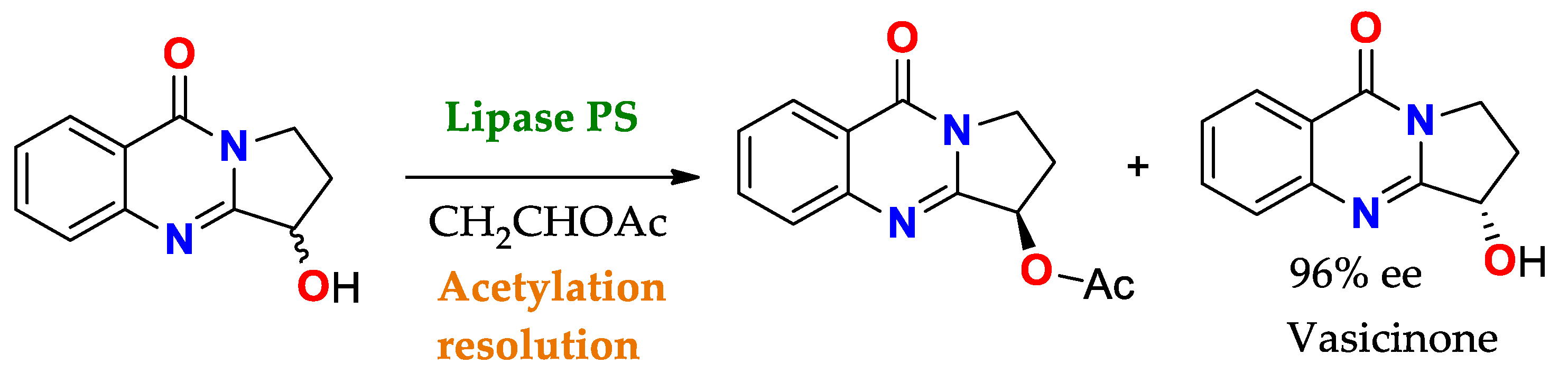
Scheme 4. Acetylation towards the resolution of the racemic mixture of 3-hydroxy-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one using PS lipase (Pseudomonas cepacia lipase) to obtain Vasicinone (molecule with bronchodilator activity and antianaphylactic action)
[86].
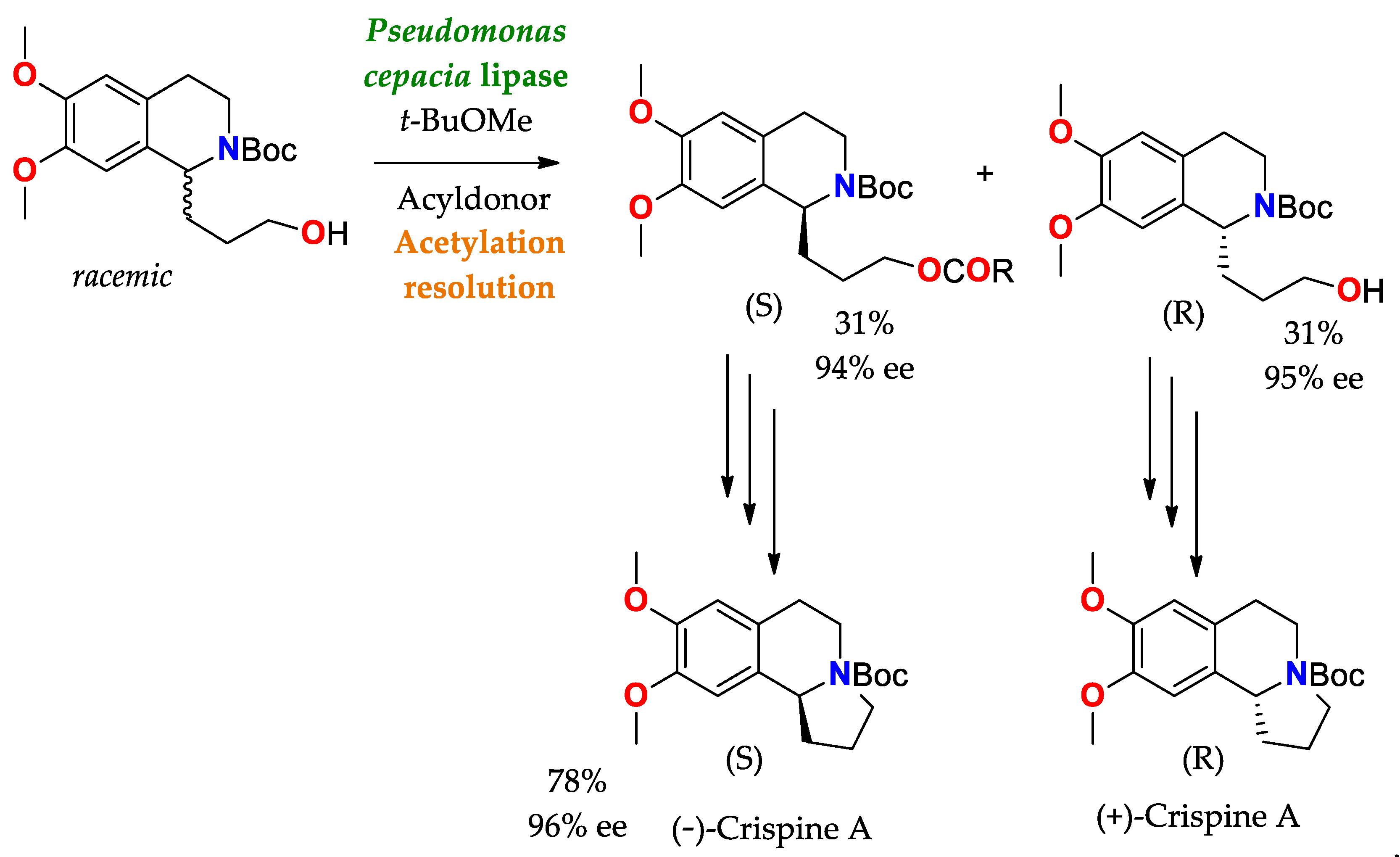
Scheme 5. Synthesis and resolution of precursors of (−)−Crispine A and (+)−Crispine A (antitumor alkaloid), by means of acetylation using a lipase as a catalyst
[87].
Scheme 6. Obtaining (S)−1−(6-bromo−2,3−difluorophenoxy)propan−2-amine by resolution using CAL-B. Levofloxacin precursor, molecule with activity as an antibiotic
[86].
Molecular Origins of the Catalytic Promiscuity of Lipases
As mentioned, due to the nature of their substrates and the interfacial mechanism of action that some lipases present, these enzymes are evolutionarily more prepared than others (such as esterases, proteases, etc.) to be functional in unconventional media and generally more hydrophobic than the aqueous medium. As Gupta indicates
[26], this particular condition means that lipases can interact with substrates of a different nature (substrate promiscuity) through interactions mediated by entropy and are therefore less specific: this is caused by the wide hydrophobic area that constitutes the catalytic domain, including the lid in some lipases. This contrasts with the interactions that occur in enzymes with smaller or more hydrophilic active centers in which polar or H bond interactions predominate and thus require more defined orientations and geometries from the substrate to form the Michaelis complex.
On the other hand, lipases, being serine hydrolases, possess an oxyanion hole that serves to polarize carbonyl groups present in their natural substrates through H bond interactions. This favors the attack of nucleophiles, including their catalytic Ser
[82]. This polarization capacity is not only limited to interacting with carbonyl groups, but also to groups such as nitrile, which will be discussed later
[88]. This oxyanion property makes it easier for the lipases to interact with substrates such as –C
α-(C=O)- R, where R’ is not only an O atom (forming an alkoxy group) but also N or S, and even C or H, as in the case of ketones and aldehydes. Particularly, in cases when the catalytic Ser is removed using site-directed mutagenesis, the catalytic His can act base activate other nucleophiles when it removes their acidic hydrogens, not only from alcohols, amines or thiols but also from C
α. The latter can be from a substrate housed in the oxyanion hole or from a substrate that is outside of it. This explains why the lipases can catalyze reactions as aldolic condensations or additions on α,β-unsaturated compounds (
Figure 7)
[82]. Significantly, lipases with these capacities are more advantageous than aldolases, as the former are more stable in non-natural conditions (they also have lower prices and broader commercial availability)
[76], which is sometimes necessary in the case of organic synthesis reactions that involve obtaining API building blocks in which the substrates are insoluble in water or anhydrous media or high temperatures are required.
 +1 credit
+1 credit