Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hai-Ying Mary Cheng | -- | 4509 | 2022-09-20 22:42:52 | | | |
| 2 | Conner Chen | Meta information modification | 4509 | 2022-09-21 11:02:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Abdalla, O.H.M.H.; Mascarenhas, B.; Cheng, H.M. Mammalian Circadian Rhythms and Ubiquitin Ligases. Encyclopedia. Available online: https://encyclopedia.pub/entry/27393 (accessed on 26 July 2026).
Abdalla OHMH, Mascarenhas B, Cheng HM. Mammalian Circadian Rhythms and Ubiquitin Ligases. Encyclopedia. Available at: https://encyclopedia.pub/entry/27393. Accessed July 26, 2026.
Abdalla, Osama Hasan Mustafa Hasan, Brittany Mascarenhas, Hai-Ying Mary Cheng. "Mammalian Circadian Rhythms and Ubiquitin Ligases" Encyclopedia, https://encyclopedia.pub/entry/27393 (accessed July 26, 2026).
Abdalla, O.H.M.H., Mascarenhas, B., & Cheng, H.M. (2022, September 20). Mammalian Circadian Rhythms and Ubiquitin Ligases. In Encyclopedia. https://encyclopedia.pub/entry/27393
Abdalla, Osama Hasan Mustafa Hasan, et al. "Mammalian Circadian Rhythms and Ubiquitin Ligases." Encyclopedia. Web. 20 September, 2022.
Copy Citation
Circadian clocks evolved to enable organisms to anticipate and prepare for periodic environmental changes driven by the day–night cycle. This internal timekeeping mechanism is built on autoregulatory transcription–translation feedback loops that control the rhythmic expression of core clock genes and their protein products. The levels of clock proteins rise and ebb throughout a 24-h period through their rhythmic synthesis and destruction. In the ubiquitin–proteasome system, the process of polyubiquitination, or the covalent attachment of a ubiquitin chain, marks a protein for degradation by the 26S proteasome. The process is regulated by E3 ubiquitin ligases, which recognize specific substrates for ubiquitination.
circadian rhythms
E3 ubiquitin ligases
ubiquitin proteasome system
1. Introduction
Life on Earth is strongly influenced by the 24-h day–night cycle, generated by the axial rotation of the planet. This cycle is accompanied by predictable environmental changes, such as daily variations in temperature, light, and food availability. Most organisms have evolved to anticipate such rhythmic occurrences in their physical environment by altering their behaviour and physiology in a similarly rhythmic fashion [1]. These biological rhythms, aptly termed circadian rhythms (from the Latin words “circa” (“approximately”) and “dies” (“a day”)), oscillate on a roughly 24-h cycle and arise from an organism’s endogenous timekeeping system, otherwise known as the circadian clock [2]. In mammals, the circadian system comprises a hierarchy of tissue-specific circadian clocks organized from the top-down and bottom-up [3][4]. The top-down arrangement occurs through the function of the master circadian pacemaker in the hypothalamus, the suprachiasmatic nucleus (SCN) [5]. As the central clock, the SCN encodes time-of-day information that it receives directly from the retina and conveys it to peripheral clocks to coordinate rhythms throughout the body [3][6]. In the bottom-up organization, peripheral clocks can feed information back to influence and regulate the activity of the SCN [3][4].
Circadian timekeeping is a cellular phenomenon that is based on transcription–translation feedback loops (TTFLs) of core clock genes and their protein products [2][7]. Through negative feedback mechanisms, TTFLs drive the rhythmic expression of core clock genes and circadian outputs with a near-24 h period. In addition to transcription, circadian rhythms are further regulated and fine-tuned by post-transcriptional, translational, post-translational, and degradative mechanisms [7].
Ubiquitination (ubiquitylation) is the process by which a small, 76-amino acid protein, ubiquitin (Ub), is conjugated onto a substrate protein, most often at a lysine residue [8][9]. A substrate may be mono-ubiquitinated (i.e., a single Ub moiety on one residue), multi-ubiquitinated (i.e., multiple residues each carrying a ubiquitin), or poly-ubiquitinated (i.e., a single residue carrying a branched or straight chain of Ub proteins) [9][10]. The specific linkages of the ubiquitin or Ub chains, and the residue that is modified, determine the fate of the substrate protein, the most studied of which is degradation by 26S proteasomes [9][10]. These are multi-protein complexes present in the cytoplasm and nucleus that recognize ubiquitylated proteins and degrade them in a processive and ATP hydrolysis-driven fashion [11]. Ubiquitination may also affect the subcellular localization of a protein or its activity and function [12].
The conjugation of ubiquitin onto a substrate and the subsequent degradation of the ubiquitinated protein are mediated by the ubiquitin proteasome system (UPS). The UPS uses a series of three protein components, termed E1, E2, and E3, to covalently attach ubiquitin on the substrate [13]. In the first step, the Ub-activating enzyme, E1, catalyzes the linkage of Ub onto an internal cysteine residue of E1 by a thioester bond [13]. This is followed by the transfer of Ub from E1 to a cysteine residue of the Ub-conjugating enzyme, E2 [13]. In the third and final step, E2 interacts with a specific E3 protein that selectively binds substrates, enabling the transfer of Ub from E2 to a lysine residue of the substrate [13]. This process can repeat to conjugate additional Ub proteins onto the previous Ub at an internal lysine residue or the N-terminal methionine (M1), generating a polyubiquitin chain [9]. Ubiquitin has seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) that are available as attachment sites for subsequent Ub proteins [9]. K48-linked chains are the most abundant linkages and direct the target protein to the 26S proteasome [9]. In humans, there are 2 genes that encode for E1, 38 for E2, and more than 600 for E3 [14]. The sheer number of E3 proteins (alternatively known as E3 ubiquitin ligases) reflects their important role in conferring substrate specificity to the UPS.
There are four major structural classes of E3 Ub ligases: the HECT (Homologous to E6-AP Carboxyl Terminus) type E3s, the RING (Really Interesting New Gene) finger type E3s, the U-box type E3s, and the RBR (RING-BetweenRING-RING) type E3s (Figure 1) [15]. These E3 ligases interact physically with E2 through their HECT, RING finger, or U-box domains [15]. In the case of HECT E3 ligases, they contain a cysteine residue that can form a thioester bond with Ub originating from the E2 protein [15]. Neither the RING finger nor the U-box E3 ligases form an intermediate with Ub; instead, they facilitate the direct transfer of Ub from E2 to the substrate [15]. With the RBR E3 ligases, E2 binds to the RING1 domain of the ligase and Ub is then directly transferred from E2 to a cysteine residue in the RING2 domain [15]. The E3 ligases of the N-end rule pathway (recently renamed the N-degron pathway) represent a special family of E3s that specifically recognize substrate proteins by their destabilizing N-terminal residue [16]. Within this family is a subclass of E3s that are defined by the presence of a UBR box, a zinc finger-like domain [16]. Most of the E3 ligases that have so far been implicated in circadian rhythms belong to the HECT and RING finger families. In addition to E3s, deubiquitinases (DUBs), which deconjugate Ub from a substrate protein, have also been shown to impact circadian rhythms by reversing the effects of ubiquitination [17][18][19].
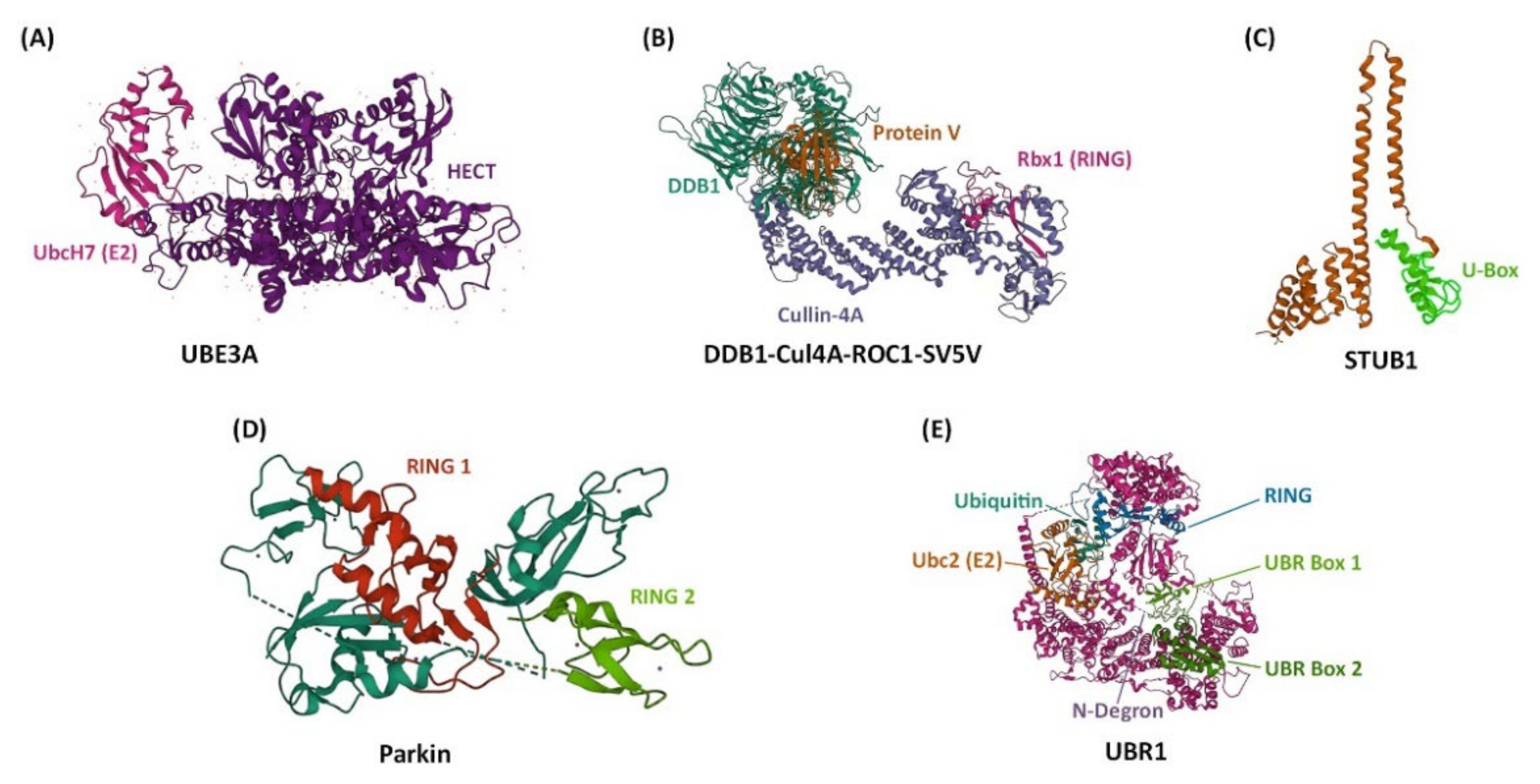
Figure 1. The structures of representative E3 ubiquitin ligases for each structural class. Atomic models were taken from the RCSB protein data bank (PDB, rcsb.org) and images were created using Mol* [20][21]. (A) UBE3A from humans, shown attached to the E2-ubiquitin-conjugating enzyme UbcH7. UBE3A is an example of a HECT type E3 ubiquitin ligase (PDB code 1C4Z) [22]. (B) DDB1–CUL4A-ROC1 from humans, shown attached to the non-structural V protein of the simian virus 5 (SV5-V). DDB1–CUL4A-ROC1 complex contains Ring-box-1 (Rbx1) and is an example of a RING finger type E3 ubiquitin ligase (PBD code 2HYE) [23]. (C) STUB1 from humans, representing a U-box type E3 ubiquitin ligase (PDB code 2C2L) [24]. (D) Parkin from brown rat, representing a RING-BetweenRING-RING type E3 ubiquitin ligase (PDB code 4K95) [25]. (E) UBR1 from yeast, shown in an initiating complex with ubiquitin, the E2-ubiquitin-conjugating enzyme Ubc2, and an N-Degron (indicated by a dashed line). All UBR proteins contain a UBR box, and UBR1 also contains a RING domain (PDB code 7MEX) [26].
2. Mammalian Circadian Rhythms and Ubiquitin Ligases
2.1. Overview of Circadian Rhythms in Mammals
2.1.1. The Suprachiasmatic Nucleus
The SCN is a bilateral structure situated in the anterior hypothalamus, dorsal to the optic chiasm [27]. It consists of approximately 20,000 neurons, each functioning as autonomous circadian clocks [28]. The SCN is subdivided into ventrolateral and dorsomedial regions, otherwise referred to as the core and shell SCN, respectively [27]. The intrinsic period of individual SCN neurons can vary from ~22 h to 30 h [28][29]. However, intercellular coupling between SCN neurons results in their oscillating in synchrony with a significantly narrower period range [28][30]. Neurons in the ventrolateral SCN receive direct inputs from the retina; they also synthesize and secrete gastrin-releasing peptide (GRP) and vasoactive intestinal polypeptide (VIP) [27]. VIP is a neuropeptide vital to interneuronal coupling within the SCN, and by extension to the robustness of the central pacemaker [31]. Once released, both VIP and GRP signal to cells in the core and shell SCN [31][32]. Dorsomedial SCN neurons utilize a different neuropeptide, arginine vasopressin (AVP), to communicate with and couple to other clock neurons [31][32]. The inhibitory neurotransmitter, gamma-aminobutyric acid (GABA), is expressed by nearly all SCN neurons and has been shown to contribute to oscillator coupling and the refinement of circadian outputs [27][33].
As the intrinsic period of the SCN deviates slightly from 24 h, photic entrainment is required to maintain synchrony between the central pacemaker and environmental cycles. This involves the daily resetting of the clock by light, which synchronizes the SCN’s phase with the solar cycle [6][34]. In mammals, photic entrainment is fully dependent on the retina, and relies on the detection of changes in environmental light intensity [34][35]. As the master pacemaker, the SCN receives and integrates environmental photic signals from the retina, using them to synchronize its own neuronal clocks, after which the SCN can convey temporal information to peripheral clocks in the brain and body through synaptic and humoral mechanisms [35]. A direct projection from the retina to the SCN, known as the retinohypothalamic tract (RHT), transmits photic signals to the central clock via the actions of secreted glutamate and pituitary adenylate cyclase-activating peptide (PACAP) [36]. Within the retina, the non-image forming, intrinsically photoreceptive retinal ganglion cells (ipRGCs) are the main players in photic entrainment, using the photopigment melanopsin to detect light in the blue wavelength range, and transmitting the photic signal to the SCN via the RHT [37][38][39].
2.1.2. The Mammalian Core Clock Machinery
Circadian TTFLs operate on the principle of negative feedback inhibition, whereby elements in the positive limb of the feedback loop drive the expression of elements within the negative limb, eventually shutting off their own expression until the cycle begins anew ~24 h later [40]. Within the primary TTFL of mammals, the core clock genes, Clock and Bmal1 (officially known as Arntl), represent the positive elements, whereas Period1 (Per1), Period2 (Per2), Cryptochrome1 (Cry1), and Cryptochrome2 (Cry2) represent the negative elements (Figure 2) [41][42][43][44][45]. The positive limb is most active during the subjective day, when the helix-loop-helix transcription factors, CLOCK and BMAL1, heterodimerize and bind to the E-box cis-regulatory elements of the Per and Cry gene promoters, activating their transcription [2][7]. Following their translation and accumulation in the cytoplasm, PER and CRY proteins heterodimerize and translocate to the nucleus [2][7]. This initiates the negative limb, as PER and CRY repress their own transcription, either by binding to CLOCK/BMAL1 and blocking E-box-mediated transactivation or by displacing CLOCK/BMAL1 from E-boxes [46]. The two modes of repression are differentially mediated by PER and CRY [46][47][48]. In the “blocking”-style repression, CRY1 binds to CLOCK-BMAL1-E-box complexes independently of PER to inhibit transactivation: the repression occurs even as CLOCK and BMAL1 remain bound to the E-box [46][47][48]. In the “displacement”-style repression, PER2, in the presence of CRY, displaces CLOCK/BMAL1 from the E-boxes [46][47][48]. PER proteins on their own have no repressive activity towards CLOCK/BMAL1 [46]. Eventually, PER and CRY are degraded by the proteasomal pathway, allowing CLOCK/BMAL1 to once again occupy E-box elements and activate gene expression [2][7].
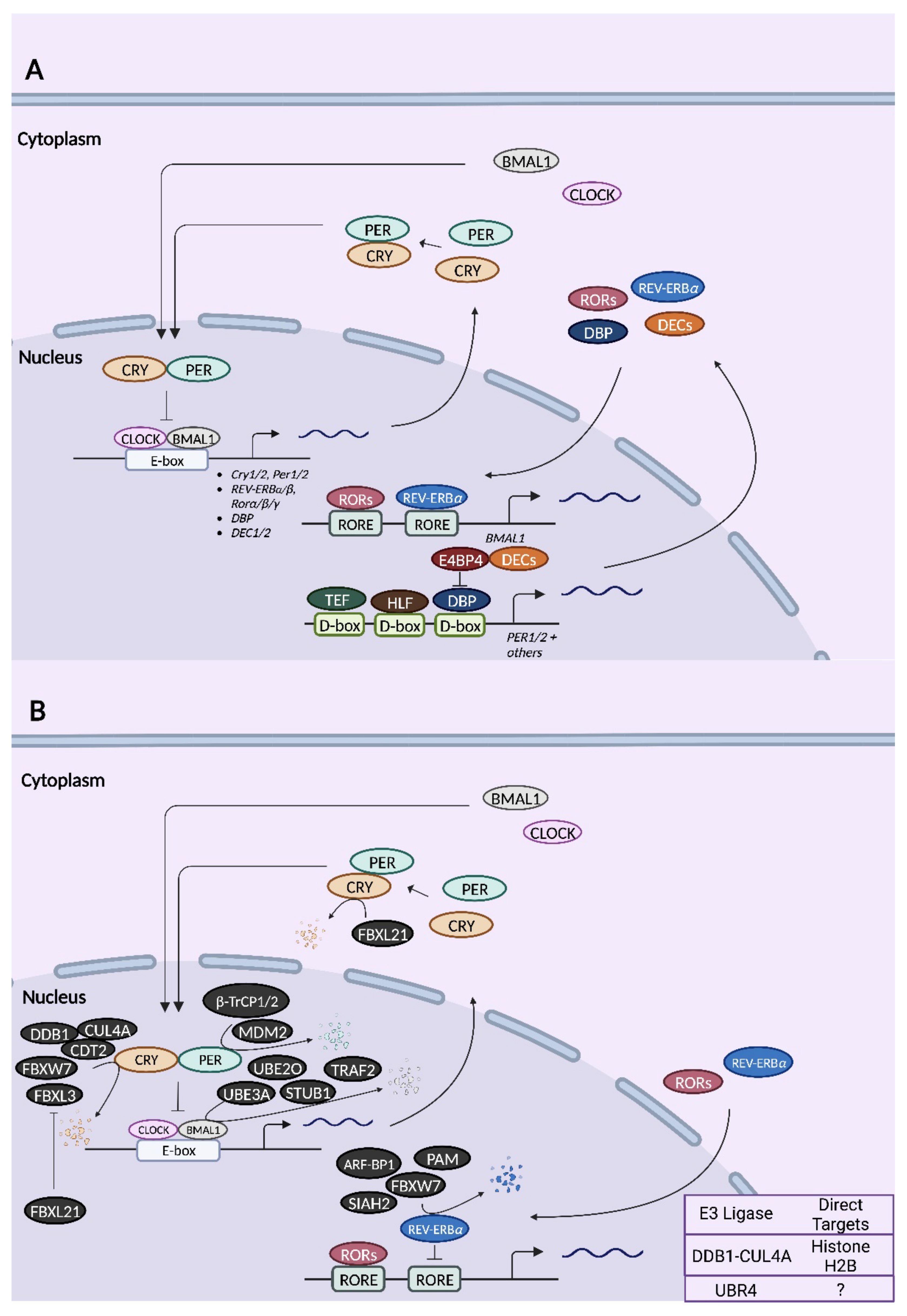
Figure 2. Schematic of mammalian circadian clock transcription–translation feedback loops (TTFLs) (A) and the relationships of known E3 ligases to core clock proteins (B). (A) In the primary TTFL of mammals, transcription factors CLOCK and BMAL1 heterodimerize and activate the transcription of target genes including Cry1/2, Per1/2, Rev-Erbα/β, Rorα/β/γ, Dbp, and Dec1/2 by binding to the E-box elements in their promoters. Once translated, PER and CRY proteins accumulate in the cytoplasm before heterodimerizing and translocating to the nucleus, where they ultimately repress E-box-dependent transcription. In a secondary TTFL, RORα/β/γ binds to ROR elements (RORE) in the promoters of Bmal1 and other genes to activate their transcription. In contrast, REV-ERBα/β binds to ROREs to repress transcription. Another auxiliary feedback loop involves DBP, TEF, and HLF, which bind to the D-box elements of Per1/2 genes (among others), initiating D-box-mediated transactivation. Binding of E4BP4 at D-box elements results in transcriptional repression. (B) All mammalian E3 ubiquitin ligases that have been demonstrated to target a core clock protein for degradation are indicated in black. E3 ligases whose targets are either unknown or not core clock proteins are indicated in the chart in the bottom right corner. Except for FBXL3 and FBXL21, the site of action (nucleus vs. cytoplasm) for all other E3 ligases is unknown. For illustrative purposes, these E3 ligases are shown as acting in the nucleus.
2.2. Mammalian Ubiquitin Ligases
2.2.1. β-TrCP1 (FBXW1) and β-TrCP2 (FBXW11)
Beta-transducin repeat-containing proteins (β-TrCP) were the first E3 ubiquitin ligases to be implicated in the regulation of the clock machinery. β-TrCP is an F-box protein of the Fbws class, characterized by the presence of an F-box motif and tandem WD40 repeats [49]. F-box proteins are the substrate recognition subunits of the SCF (Skp1-Cullin 1-F-box protein) family of E3 ubiquitin ligases [49]. Casein kinase 1 (CK1)-mediated phosphorylation of PER2 was shown to trigger an association between β-TrCP and PER2 [50]. The interaction between β-TrCP and PER2 is also facilitated by the SUMOylation of PER2 by SUMO2 [51]. The expression of mutant β-TrCP that lacks an F-box inhibits the degradation of phosphorylated PER2 [50]. In another study, β-TrCP1 and β-TrCP2 were identified as binding partners of PER1 [52]. These interactions are dependent on the phosphorylation of PER1 by CK1ε [52]. Furthermore, the silencing of β-TrCP1 stabilizes PER1 and inhibits CK1ε-induced PER1 degradation [52]. Cell-free assays showed that SCF complexes containing β-TrCP are capable of ubiquitinating PER1 [52]. In NIH-3T3 fibroblasts, the knockdown of β-TrCP1 or the expression of dominant-negative β-TrCP1 elicits a lengthening of molecular oscillations [53]. PER2 mutants that are unable to interact physically with β-TrCP1/2 exhibit severely disrupted or damped rhythms in fibroblasts [53]. However, β-TrCP1 appears to be dispensable for circadian rhythms at the behavioural level, as β-TrCP1 knockout mice are phenotypically normal with respect to period length and light-induced phase shifts [54]. On the other hand, introducing the PER2 S478A mutation, which can no longer be phosphorylated by CK1δ/ε and thus cannot recruit β-TrCP1/2, results in a dramatic lengthening of the behavioural period in knock-in mice and the accumulation of PER2 protein in the nucleus and cytoplasm of the liver, suggesting that β-TrCP2 may compensate for the loss of β-TrCP1 [55]. In line with this, inducible β-TrCP2 knockout mice exhibit a dramatic circadian phenotype characterized by unstable behavioural rhythms and period variability under constant darkness (DD) [56]. The fact that the ubiquitination of PER2 is still observed in the absence of β-TrCP1 and β-TrCP2 indicates that PER2 is a substrate of other E3 ligases [56]. β-TrCP1/2 may also influence the circadian clock by ubiquitinating other clock proteins such as DEC1 and targeting them for proteasomal degradation [57].
2.2.2. Mouse Double Minute 2 Homolog (MDM2)
MDM2 is a RING finger type E3 ligase that serves as a scaffold, bringing E2 enzymes to protein substrates for ubiquitination [58]. PER2 can form trimeric complexes with MDM2 and its substrate, p53 [59]. However, PER2 and MDM2 can directly associate with each other independently of p53 [59]. Furthermore, MDM2 binds to PER2 at the latter’s PAS domain and an inner region that undergoes extensive phosphorylation [59]. CK1δ/ε-mediated phosphorylation of PER2 is not necessary for either MDM2 binding or MDM2-dependent ubiquitination of PER2 [59]. MDM2 destabilizes PER2 and appears to work cooperatively with β-TrCP to control the abundance of PER2 during the rising and falling phases of the protein’s circadian cycle [59]. The knockdown of mdm2 extends the circadian period in murine embryonic fibroblasts (MEFs), whereas the overexpression of mdm2 shortens it [59].
2.2.3. FBXL3
FBXL3 was first reported in three sister studies in 2007 as an E3 ubiquitin ligase for CRY proteins [60][61][62]. By chemical-induced mutagenesis, two mutations in FBXL3 were identified that dramatically lengthen the circadian period: the C358S substitution termed afterhours (Afh) and the I364T mutation termed overtime (Ovtm) [61][62]. In both Afh and Ovtm mutant mice, CRY1/2 protein levels are not appreciably altered but PER1/2 abundance is strongly suppressed, suggesting that the stabilization of CRY1/2 in the presence of lower E3 ligase activity is compensated for by a reduction in E-box-dependent transcription of Cry1/2 genes [61][62]. FBXL3 binds specifically to CRY1 and CRY2 but not to other core clock proteins and promotes their degradation by proteasomes [60][62]. FBXL3 was also shown to promote CRY2 ubiquitination in an F-box-dependent manner, suggesting a direct effect of FBXL3 on CRY2 [60]. Both the Afh and Ovtm mutations reduce the rate of CRY1/2 degradation; in the case of the Afh mutation, this was attributed to the reduced binding of FBXL3(Afh) to CRY proteins and reduced catalytic activity [60][62]. As a consequence of their effects on CRY stability, these mutations dampen the amplitude of circadian gene expression [61][62].
Crystal structure analysis revealed a bipartite interaction between FBXL3 and CRY2, in which the C-terminal tail of FBXL3 occupies the FAD-binding pocket of CRY2, and the leucine-rich repeat (LRR) domain of FBXL3 is a secondary site of contact for CRY2 at three key structural motifs [63]. As these interactions occur in the absence of FAD or PER binding to CRY2, either of these factors can disrupt the FBXL3–CRY2 complex or prevent its formation [63]. The formation of FBXL3–CRY complexes is required for the recruitment of SKP1 and CUL1, thereby forming the fully functional SCF complex [64]. This substrate-dependent formation of SCF complexes appears to be specific for FBXL3 and is not observed with FBXL21 [64].
CRY2 binding may also recruit FBXL3 to other substrates such as c-MYC and TLK2 to induce their ubiquitination and subsequent degradation, thereby linking the clock to proteolysis in other physiological systems [65]. In addition to CRY1/2, FBXL3 has also been shown to physically associate with REV-ERBα in mouse livers [66]. REV-ERBα recruits FBXL3 to RORE sites, where it derepresses gene expression by inhibiting the actions of REV-ERBα:HDAC3 complexes [66].
2.2.4. FBXL21
The F-box protein FBXL21 is the closest homologue of FBXL3 [49]. Initial studies revealed that FBXL21 is expressed in the brain and neuroendocrine tissues of sheep, and physically associates with ovine CRY1 [67]. Fbxl21 harbours functional E- and D-box elements within its promoter, resulting in high and rhythmic expression of the gene in the ovine and murine SCN [67]. Furthermore, FBXL21 overexpression abrogates the repressive effects of CRY1 on CLOCK/BMAL1-mediated transcription [67]. A subsequent study showed that the past-time (Psttm) mutation, which is a missense mutation (G149E) in the Fbxl21 gene, significantly shortens the circadian period and antagonizes the period-lengthening effects of FBXL3(Ovtm) [68]. FBXL21 binds to CRY1/2 with a higher affinity than FBXL3, effectively outcompeting FBXL3 [68]. In the nucleus, where both FBXL21 and FBXL3 are present, FBXL21 sequesters CRY proteins from FBXL3 and protects them from FBXL3-induced proteasomal degradation [68]. In the cytosol, where FBXL3 is absent, FBXL21 triggers the slow degradation of CRY1/2 [68]. It was further shown that FBXL21 preferentially forms SCF complexes in the cytoplasm but not in the nucleus [68]. These collective observations are consistent with the different potencies of the two FBXL homologs in CRY destabilization, where FBXL21 appears to be a weaker E3 ligase for CRY than FBXL3 [68]. The effects of FBXL21 on the stability of nuclear and cytoplasmic CRY are such that Psttm mutant mice have altered core clock gene expression that is characterized by a higher expression of E-box-regulated genes [68]. Although a different group confirmed many of these findings, including the antagonism between FBXL3 and FBXL21, the stabilization of CRY by FBXL21, and the preferential localization of FBXL21 in the cytoplasm, in their case, Fbxl21-deficient mice did not show a circadian period phenotype, unlike the Psttm mutants [69].
2.2.5. DDB1–CUL4A and DDB1–CUL4A–CDT2
The DDB1–CUL4A–CDT2 E3 ubiquitin ligase complex has been shown to target CRY1 for degradation [70][71]. In vitro assays revealed that DDB1–CUL4A–CDT2 directly ubiquitinates CRY1 at Lys-585, marking the protein for proteasomal degradation [71]. CRY1 physically binds to CDT2 and the silencing of Cdt2 prevents complex formation between CRY1 and DDB1–CUL4A [71]. In mouse hepatoma cells, the knockdown of Ddb1 or overexpression of the ubiquitination-defective mutant CRY1 K585A enhances CRY1 stability and increases the amplitude of circadian oscillations as measured by a Bmal1-Luc reporter [71]. In a subsequent study, liver-specific Ddb1 knockout mice were shown to have impairments in hepatic gluconeogenesis but were protected from high-fat-diet-induced hyperglycemia [70]. These effects are due to elevated levels of CRY1, which binds to the FOXO1 transcription factor and promotes its ubiquitination and degradation [70]. In turn, the lower abundance of FOXO1 in Ddb1 knockouts suppresses gluconeogenic gene expression [70]. Besides CRY1, DDB1–CUL4A has been shown to interact with CLOCK-BMAL1 [72]. These circadian transcription factors bind to an adaptor protein of the DDB1–CUL4A complex, WD repeat-containing protein 76 (WDR76) [72]. Through this interaction, CLOCK-BMAL1 recruits DDB1–CUL4A to E-boxes of the Per1 and Per2 genes as well as other circadian genes [72]. DDB1–CUL4A enhances the monoubiquitination of histone H2B at E-box sites, which subsequently inhibits CLOCK-BMAL1 binding while promoting the association with PER complexes [72].
2.2.6. FBXW7
FBXW7 is an F-box protein of the Fbws class [49]. Several studies have implicated the involvement of FBXW7 in the regulation of circadian rhythms by targeting different proteins. In mice injected with renal carcinoma cells, the expression of FBXW7 exhibits circadian oscillations in the tumours, driven by DBP, which binds to and transactivates Fbxw7 in a rhythmic fashion [73]. The mammalian target of rapamycin (mTOR), a key protein in cell growth and the circadian control of translation, oscillates in anti-phase to FBXW7 protein [73]. A prior study demonstrated that FBXW7 ubiquitinates mTOR and targets it for proteasomal degradation [74]. A separate study revealed that FBXW7 binds to CRY2 in colorectal cancer cells, potentially through a direct interaction between the degron motif of CRY2 and the narrow face of the WD40 domain of FBXW7 [75]. It was further shown that FBXW7 destabilizes CRY2 by promoting its ubiquitination and subsequent proteasomal degradation [75]. REV-ERBα is another identified target of FBXW7 [76]. FBXW7 was demonstrated to physically interact with REV-ERBα, enhancing its ubiquitination and destabilizing it [76]. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of REV-ERBα at Thr-275 is required for its recognition by FBXW7 [76]. The amplitude of circadian gene expression is suppressed when Fbxw7 is silenced in cultured cells or ablated in mouse livers [76]. The deletion of Fbxw7 specifically in the liver alters the hepatic circadian transcriptome and disrupts whole-body lipid and glucose metabolism [76].
2.2.7. TNF Receptor-Associated Factor 2 (TRAF2)
The ubiquitin ligase TRAF2 was initially identified as a CRY1-binding protein in a high-throughput yeast two-hybrid screen [77]. However, a subsequent study showed that TRAF2 overexpression does not alter CRY1 abundance, suggesting that the interaction between TRAF2 and CRY1 does not lead to the degradation of the latter [78]. The same study also revealed that TRAF2 physically binds to BMAL1 and reduces its abundance [78]. The physical association is mediated by the zinc finger domain of TRAF2 and not by the TRAF domain, the canonical substrate recognition site [78]. Furthermore, the deletion of the RING domain of TRAF2 stabilizes BMAL1 protein, indicating that the effects of TRAF2 are dependent on its ubiquitin ligase activity [78]. Consistent with these results, the overexpression of TRAF2 promotes the ubiquitination of BMAL1 and its degradation by proteasomes [78]. The TRAF2-dependent reduction in BMAL1 abundance ultimately attenuates E-box-mediated transcription and dampens Per1 oscillations [78].
2.2.8. STIP1 Homology and U-Box-Containing Protein 1 (STUB1)
STUB1 was identified in a mass spectrometric analysis of BMAL1-binding partners [79]. This interaction is selective for BMAL1 and is not observed with CLOCK [79]. The overexpression of wild-type STUB1, but not of a catalytically inactive form, reduces the abundance of BMAL1 protein in HEK293T cells, indicating that STUB1 affects BMAL1 stability through its ubiquitin ligase activity [79]. Along these lines, STUB1 catalyzes the K48-linked polyubiquitination of BMAL1, which is associated with proteasomal degradation [79]. STUB1 is primarily localized to the cytosol, but upon oxidative stress, it translocates to the nucleus where it can destabilize BMAL1 to attenuate cellular senescence [79].
2.2.9. UBE3A
UBE3A is a HECT-domain-containing ubiquitin ligase expressed in multiple tissues, including the SCN [80]. It is also the causative gene for the neurodevelopmental disorder Angelman Syndrome (AS), in which UBE3A expression is absent due to the loss of the maternal allele amid the normally silenced paternal allele [81]. There is evidence for and against paternal imprinting of the Ube3a gene in SCN neurons [82][83]. Sleep disturbances are one of the symptoms of AS, which include delayed development, intellectual disability, impaired speech, and motor dysfunction [84]. Ablating the maternal copy of Ube3a in mice consistently disrupts sleep homeostasis [80][83][85]. However, one study found that this mutation also lengthens the circadian period under constant darkness (DD), accelerates recovery from jetlag (i.e., mice re-entrain more rapidly to an advanced light–dark schedule), and suppresses locomotor activity under constant light (LL), whereas another study found no effect on circadian period [80][86]. At the molecular level, the activation of UBE3A in mouse embryonic fibroblasts (MEFs) by the viral oncogenes E6/E7 triggers the ubiquitination of BMAL1 and a reduction in its protein abundance through proteasomal degradation, ultimately leading to a loss of circadian rhythms [87]. These effects are direct, as UBE3A physically binds to, polyubiquitinates, and destabilizes BMAL1 [86][87]. Consistent with these observations, the loss of the maternal allele of Ube3a elevates BMAL1 abundance in the murine hypothalamus [86]. Lastly, even in the absence of E6/E7-mediated transformation, the endogenous activity of UBE3A is required for maintaining robust rhythms of Per2 expression in MEFs [87].
2.2.10. UBE2O
UBE2O is a ubiquitin-conjugating enzyme with hybrid E2/E3 activity [88]. Initially identified from a mass spectrometry screen for BMAL1-binding proteins, UBE2O was shown to physically associate with BMAL1 in mouse Neuro2a cells and whole brain tissue [89]. The overexpression of UBE2O reduces the levels of endogenous BMAL1 in HEK293T cells in a dose-dependent fashion, whereas its knockdown elevates BMAL1 abundance [89]. These effects are specific to BMAL1 and are not observed with CLOCK [89]. UBE2O was further shown to ubiquitinate BMAL1 and dramatically reduce its half-life [89]. The conserved region 2 (CR2) domain of UBE2O is essential for BMAL1 binding and ubiquitination [89]. The effects of UBE2O on BMAL1 stability lead to attenuated BMAL1 transcriptional activity when UBE2O is overexpressed and a higher amplitude of Per2 rhythms when it is silenced [89].
2.2.11. Seven in Absentia 2 (SIAH2)
SIAH2 was identified in a functional screen for REV-ERBα-directed ubiquitin ligases [90]. The overexpression of SIAH2 selectively destabilizes REV-ERBα and REV-ERBβ, but not the other proteins tested, including PER1, PER2, CRY1, and CLOCK [90]. In contrast, the SIAH2 paralog, SIAH1, does not influence REV-ERB stability [90]. More importantly, SIAH2 was shown to physically interact with REV-ERBα/β and promote its ubiquitination [90]. Ablating the RING domain of SIAH2 interferes with its ability to destabilize REV-ERB, indicating the importance of its catalytic function as an E3 ubiquitin ligase [90]. In synchronized U2OS cells, the knockdown of Siah2 slows the turnover of REV-ERBα, thereby affecting its rhythms and the expression of its target genes, as well as lengthening the circadian period [90]. However, in mouse models, the absence of Siah2 does not impact REV-ERBα protein rhythms, likely as a result of compensation by other E3 ligases, although other clock genes such as Bmal1 and Per2 are moderately affected at the transcript level [91]. In a surprising twist, Siah2 deficiency disrupts the circadian hepatic transcriptome only in female mice [91]. Global circadian gene expression in the female liver is phase-advanced by ~9 h such that genes that are normally expressed during the night now peak in the daytime [91]. The underlying cause for this sexual dimorphism remains unknown.
2.2.12. ARF-BP1 and PAM (Myc-BP2)
ARF-BP1 (also known as HUWE1) and PAM are HECT and RING finger type E3 ligases, respectively [92][93]. Both were co-purified with REV-ERBα in the presence of lithium chloride, an inhibitor of glycogen synthase kinase 3 beta (GSK3β) and a known inducer of REV-ERBα degradation, and subsequently identified by mass spectrometry [94]. Co-overexpression of ARF-BP1 and PAM strongly suppresses REV-ERBα protein levels, whereas their simultaneous depletion stabilizes REV-ERBα [94]. Furthermore, ARF-BP1 and PAM exclusively promote the K48-linked polyubiquitination of REV-ERBα, targeting the protein for degradation [94]. The knockdown of both ARF-BP1 and PAM in hepatoma cells results in a higher amplitude of REV-ERBα protein rhythms and consequently a lower expression and oscillatory amplitude of Bmal1 [94].
References
- Moore-Ede, M.C. Physiology of the Circadian Timing System: Predictive versus Reactive Homeostasis. Am. J. Physiol. 1986, 250, R737–R752.
- Lowrey, P.L.; Takahashi, J.S. Chapter 6—Genetics of Circadian Rhythms in Mammalian Model Organisms. In The Genetics of Circadian Rhythms; Brody, S., Ed.; Advances in Genetics; Academic Press: Cambridge, MA, USA, 2011; Volume 74, pp. 175–230.
- Buijs, F.N.; León-Mercado, L.; Guzmán-Ruiz, M.; Guerrero-Vargas, N.N.; Romo-Nava, F.; Buijs, R.M. The Circadian System: A Regulatory Feedback Network of Periphery and Brain. Physiology 2016, 31, 170–181.
- Roenneberg, T.; Merrow, M. The Circadian Clock and Human Health. Curr. Biol. 2016, 26, R432–R443.
- Ralph, M.R.; Foster, R.G.; Davis, F.C.; Menaker, M. Transplanted Suprachiasmatic Nucleus Determines Circadian Period. Science 1990, 247, 975–978.
- Golombek, D.A.; Rosenstein, R.E. Physiology of Circadian Entrainment. Physiol. Rev. 2010, 90, 1063–1102.
- Mendoza-Viveros, L.; Bouchard-Cannon, P.; Hegazi, S.; Cheng, A.H.; Pastore, S.; Cheng, H.-Y.M. Molecular Modulators of the Circadian Clock: Lessons from Flies and Mice. Cell. Mol. Life Sci. 2017, 74, 1035–1059.
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular Functions and Molecular Mechanisms of Non-Lysine Ubiquitination. Open Biol. 2019, 9, 190147.
- Yau, R.; Rape, M. The Increasing Complexity of the Ubiquitin Code. Nat. Cell Biol. 2016, 18, 579–586.
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886.
- Baumeister, W.; Walz, J.; Zühl, F.; Seemüller, E. The Proteasome: Paradigm of a Self-Compartmentalizing Protease. Cell 1998, 92, 367–380.
- Liao, Y.; Sumara, I.; Pangou, E. Non-Proteolytic Ubiquitylation in Cellular Signaling and Human Disease. Commun. Biol. 2022, 5, 114.
- Kleiger, G.; Mayor, T. Perilous Journey: A Tour of the Ubiquitin-Proteasome System. Trends Cell Biol. 2014, 24, 352–359.
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as Attractive Anti-Cancer Targets. Curr. Pharm. Des. 2013, 19, 3215–3225.
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 Ubiquitin Ligases: Styles, Structures and Functions. Mol. Biomed. 2021, 2, 23.
- Tasaki, T.; Sriram, S.M.; Park, K.S.; Kwon, Y.T. The N-End Rule Pathway. Annu. Rev. Biochem. 2012, 81, 261–289.
- Yang, Y.; Duguay, D.; Bédard, N.; Rachalski, A.; Baquiran, G.; Na, C.H.; Fahrenkrug, J.; Storch, K.-F.; Peng, J.; Wing, S.S.; et al. Regulation of Behavioral Circadian Rhythms and Clock Protein PER1 by the Deubiquitinating Enzyme USP2. Biol. Open 2012, 1, 789–801.
- Yang, Y.; Duguay, D.; Fahrenkrug, J.; Cermakian, N.; Wing, S.S. USP2 Regulates the Intracellular Localization of PER1 and Circadian Gene Expression. J. Biol. Rhythms 2014, 29, 243–256.
- Eletr, Z.M.; Wilkinson, K.D. Regulation of Proteolysis by Human Deubiquitinating Enzymes. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 114–128.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242.
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437.
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2–E3 Enzyme Cascade. Science 1999, 286, 1321–1326.
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular Architecture and Assembly of the DDB1–CUL4A Ubiquitin Ligase Machinery. Nature 2006, 443, 590–593.
- Zhang, M.; Windheim, M.; Roe, S.M.; Peggie, M.; Cohen, P.; Prodromou, C.; Pearl, L.H. Chaperoned Ubiquitylation—Crystal Structures of the CHIP U Box E3 Ubiquitin Ligase and a CHIP-Ubc13-Uev1a Complex. Mol. Cell 2005, 20, 525–538.
- Trempe, J.-F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science 2013, 340, 1451–1455.
- Pan, M.; Zheng, Q.; Wang, T.; Liang, L.; Mao, J.; Zuo, C.; Ding, R.; Ai, H.; Xie, Y.; Si, D.; et al. Structural Insights into Ubr1-Mediated N-Degron Polyubiquitination. Nature 2021, 600, 334–338.
- Abrahamson, E.E.; Moore, R.Y. Suprachiasmatic Nucleus in the Mouse: Retinal Innervation, Intrinsic Organization and Efferent Projections. Brain Res. 2001, 916, 172–191.
- Herzog, E.D.; Takahashi, J.S.; Block, G.D. Clock Controls Circadian Period in Isolated Suprachiasmatic Nucleus Neurons. Nat. Neurosci. 1998, 1, 708–713.
- Welsh, D.K.; Logothetis, D.E.; Meister, M.; Reppert, S.M. Individual Neurons Dissociated from Rat Suprachiasmatic Nucleus Express Independently Phased Circadian Firing Rhythms. Neuron 1995, 14, 697–706.
- Yamaguchi, S.; Isejima, H.; Matsuo, T.; Okura, R.; Yagita, K.; Kobayashi, M.; Okamura, H. Synchronization of Cellular Clocks in the Suprachiasmatic Nucleus. Science 2003, 302, 1408–1412.
- Maywood, E.S.; Chesham, J.E.; Brien, J.A.; Hastings, M.H. A Diversity of Paracrine Signals Sustains Molecular Circadian Cycling in Suprachiasmatic Nucleus Circuits. Proc. Natl. Acad. Sci. USA 2011, 108, 14306–14311.
- Varadarajan, S.; Tajiri, M.; Jain, R.; Holt, R.; Ahmed, Q.; LeSauter, J.; Silver, R. Connectome of the Suprachiasmatic Nucleus: New Evidence of the Core-Shell Relationship. eNeuro 2018, 5, ENEURO.0205-18.2018.
- Ono, D.; Honma, K.; Yanagawa, Y.; Yamanaka, A.; Honma, S. GABA in the Suprachiasmatic Nucleus Refines Circadian Output Rhythms in Mice. Commun. Biol. 2019, 2, 232.
- Ashton, A.; Foster, R.G.; Jagannath, A. Photic Entrainment of the Circadian System. Int. J. Mol. Sci. 2022, 23, 729.
- Hughes, S.; Jagannath, A.; Hankins, M.W.; Foster, R.G.; Peirson, S.N. Chapter Six—Photic Regulation of Clock Systems. In Methods in Enzymology; Sehgal, A., Ed.; Circadian Rhythms and Biological Clocks, Part B; Academic Press: Cambridge, MA, USA, 2015; Volume 552, pp. 125–143.
- Hannibal, J. Neurotransmitters of the Retino-Hypothalamic Tract. Cell Tissue Res. 2002, 309, 73–88.
- Berson, D.M.; Dunn, F.A.; Takao, M. Phototransduction by Retinal Ganglion Cells That Set the Circadian Clock. Science 2002, 295, 1070–1073.
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-Containing Retinal Ganglion Cells: Architecture, Projections, and Intrinsic Photosensitivity. Science 2002, 295, 1065–1070.
- Provencio, I.; Rollag, M.D.; Castrucci, A.M. Photoreceptive Net in the Mammalian Retina. This Mesh of Cells May Explain How Some Blind Mice Can Still Tell Day from Night. Nature 2002, 415, 493.
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and Peripheral Circadian Clocks in Mammals. Annu. Rev. Neurosci. 2012, 35, 445–462.
- Bunger, M.K.; Wilsbacher, L.D.; Moran, S.M.; Clendenin, C.; Radcliffe, L.A.; Hogenesch, J.B.; Simon, M.C.; Takahashi, J.S.; Bradfield, C.A. Mop3 Is an Essential Component of the Master Circadian Pacemaker in Mammals. Cell 2000, 103, 1009–1017.
- Darlington, T.K.; Wager-Smith, K.; Ceriani, M.F.; Staknis, D.; Gekakis, N.; Steeves, T.D.; Weitz, C.J.; Takahashi, J.S.; Kay, S.A. Closing the Circadian Loop: CLOCK-Induced Transcription of Its Own Inhibitors Per and Tim. Science 1998, 280, 1599–1603.
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK Protein in the Mammalian Circadian Mechanism. Science 1998, 280, 1564–1569.
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.R.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 Are Essential Components of the Negative Limb of the Circadian Clock Feedback Loop. Cell 1999, 98, 193–205.
- Patke, A.; Young, M.W.; Axelrod, S. Molecular Mechanisms and Physiological Importance of Circadian Rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84.
- Ye, R.; Selby, C.P.; Chiou, Y.-Y.; Ozkan-Dagliyan, I.; Gaddameedhi, S.; Sancar, A. Dual Modes of CLOCK:BMAL1 Inhibition Mediated by Cryptochrome and Period Proteins in the Mammalian Circadian Clock. Genes Dev. 2014, 28, 1989–1998.
- Ye, R.; Selby, C.P.; Ozturk, N.; Annayev, Y.; Sancar, A. Biochemical Analysis of the Canonical Model for the Mammalian Circadian Clock. J. Biol. Chem. 2011, 286, 25891–25902.
- Cao, X.; Yang, Y.; Selby, C.P.; Liu, Z.; Sancar, A. Molecular Mechanism of the Repressive Phase of the Mammalian Circadian Clock. Proc. Natl. Acad. Sci. USA 2021, 118, e2021174118.
- Jin, J.; Cardozo, T.; Lovering, R.C.; Elledge, S.J.; Pagano, M.; Harper, J.W. Systematic Analysis and Nomenclature of Mammalian F-Box Proteins. Genes Dev. 2004, 18, 2573–2580.
- Eide, E.; Margaret, W.; Heeseog, K.; Peter, W.; William, H.; Fernando, C.; Erica, V.; Andrew, G.; David, V. Control of Mammalian Circadian Rhythm by CKIepsilon-Regulated Proteasome-Mediated PER2 Degradation. Mol. Cell. Biol. 2005, 25, 2795–2807.
- Chen, L.-C.; Hsieh, Y.-L.; Tan, G.Y.T.; Kuo, T.-Y.; Chou, Y.-C.; Hsu, P.-H.; Hwang-Verslues, W.W. Differential Effects of SUMO1 and SUMO2 on Circadian Protein PER2 Stability and Function. Sci. Rep. 2021, 11, 14431.
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCFβ-TRCP Controls Clock-Dependent Transcription via Casein Kinase 1-Dependent Degradation of the Mammalian Period-1 (Per1) Protein. J. Biol. Chem. 2005, 280, 26863–26872.
- Reischl, S.; Vanselow, K.; Westermark, P.O.; Thierfelder, N.; Maier, B.; Herzel, H.; Kramer, A. β-TrCP1-Mediated Degradation of PERIOD2 Is Essential for Circadian Dynamics. J. Biol. Rhythms 2007, 22, 375–386.
- Ohsaki, K.; Oishi, K.; Kozono, Y.; Nakayama, K.; Nakayama, K.I.; Ishida, N. The Role of β-TrCP1 and β-TrCP2 in Circadian Rhythm Generation by Mediating Degradation of Clock Protein PER2. J. Biochem. 2008, 144, 609–618.
- Masuda, S.; Narasimamurthy, R.; Yoshitane, H.; Kim, J.K.; Fukada, Y.; Virshup, D.M. Mutation of a PER2 Phosphodegron Perturbs the Circadian Phosphoswitch. Proc. Natl. Acad. Sci. USA 2020, 117, 10888–10896.
- D’Alessandro, M.; Beesley, S.; Kim, J.K.; Jones, Z.; Chen, R.; Wi, J.; Kyle, K.; Vera, D.; Pagano, M.; Nowakowski, R.; et al. Stability of Wake-Sleep Cycles Requires Robust Degradation of the PERIOD Protein. Curr. Biol. 2017, 27, 3454–3467.
- Kim, J.; D’Annibale, S.; Magliozzi, R.; Low, T.Y.; Jansen, P.; Shaltiel, I.A.; Mohammed, S.; Heck, A.J.R.; Medema, R.H.; Guardavaccaro, D. USP17- and SCFβTrCP-Regulated Degradation of DEC1 Controls the DNA Damage Response. Mol. Cell. Biol. 2014, 34, 4177–4185.
- Honda, R.; Yasuda, H. Activity of MDM2, a Ubiquitin Ligase, toward P53 or Itself Is Dependent on the RING Finger Domain of the Ligase. Oncogene 2000, 19, 1473–1476.
- Liu, J.; Zou, X.; Gotoh, T.; Brown, A.M.; Jiang, L.; Wisdom, E.L.; Kim, J.K.; Finkielstein, C. V Distinct Control of PERIOD2 Degradation and Circadian Rhythms by the Oncoprotein and Ubiquitin Ligase MDM2. Sci. Signal. 2018, 11, eaau0715.
- Busino, L.; Bassermann, F.; Maiolica, A.; Lee, C.; Nolan, P.M.; Godinho, S.I.H.; Draetta, G.F.; Pagano, M. SCFFbxl3 Controls the Oscillation of the Circadian Clock by Directing the Degradation of Cryptochrome Proteins. Science 2007, 316, 900–904.
- Godinho, S.I.H.; Maywood, E.S.; Shaw, L.; Tucci, V.; Barnard, A.R.; Busino, L.; Pagano, M.; Kendall, R.; Quwailid, M.M.; Romero, M.R.; et al. The After-Hours Mutant Reveals a Role for Fbxl3 in Determining Mammalian Circadian Period. Science 2007, 316, 897–900.
- Siepka, S.M.; Yoo, S.-H.; Park, J.; Song, W.; Kumar, V.; Hu, Y.; Lee, C.; Takahashi, J.S. Circadian Mutant Overtime Reveals F-Box Protein FBXL3 Regulation of Cryptochrome and Period Gene Expression. Cell 2007, 129, 1011–1023.
- Xing, W.; Busino, L.; Hinds, T.R.; Marionni, S.T.; Saifee, N.H.; Bush, M.F.; Pagano, M.; Zheng, N. SCFFBXL3 Ubiquitin Ligase Targets Cryptochromes at Their Cofactor Pocket. Nature 2013, 496, 64–68.
- Yumimoto, K.; Muneoka, T.; Tsuboi, T.; Nakayama, K.I. Substrate Binding Promotes Formation of the Skp1-Cul1-Fbxl3 (SCFFbxl3) Protein Complex. J. Biol. Chem. 2013, 288, 32766–32776.
- Correia, S.; Chan, A.; Vaughan, M.; Zolboot, N.; Perea, V.; Huber, A.-L.; Kriebs, A.; Moresco, J.; Yates, J.; Lamia, K. The Circadian E3 Ligase Complex SCFFBXL3+CRY Targets TLK2. Sci. Rep. 2019, 9, 198.
- Shi, G.; Xing, L.; Liu, Z.; Qu, Z.; Wu, X.; Dong, Z.; Wang, X.; Gao, X.; Huang, M.; Yan, J.; et al. Dual Roles of FBXL3 in the Mammalian Circadian Feedback Loops Are Important for Period Determination and Robustness of the Clock. Proc. Natl. Acad. Sci. USA 2013, 110, 4750–4755.
- Dardente, H.; Mendoza, J.; Fustin, J.-M.; Challet, E.; Hazlerigg, D.G. Implication of the F-Box Protein FBXL21 in Circadian Pacemaker Function in Mammals. PLoS ONE 2008, 3, e3530.
- Yoo, S.-H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.-K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 Ubiquitin Ligases Govern Circadian Periodicity by Degradation of CRY in Nucleus and Cytoplasm. Cell 2013, 152, 1091–1105.
- Hirano, A.; Yumimoto, K.; Tsunematsu, R.; Matsumoto, M.; Oyama, M.; Kozuka-Hata, H.; Nakagawa, T.; Lanjakornsiripan, D.; Nakayama, K.I.; Fukada, Y. FBXL21 Regulates Oscillation of the Circadian Clock through Ubiquitination and Stabilization of Cryptochromes. Cell 2013, 152, 1106–1118.
- Tong, X.; Zhang, D.; Charney, N.; Jin, E.; VanDommelen, K.; Stamper, K.; Gupta, N.; Saldate, J.; Yin, L. DDB1-Mediated CRY1 Degradation Promotes FOXO1-Driven Gluconeogenesis in Liver. Diabetes 2017, 66, 2571–2582.
- Tong, X.; Zhang, D.; Guha, A.; Arthurs, B.; Cazares, V.; Gupta, N.; Yin, L. CUL4-DDB1-CDT2 E3 Ligase Regulates the Molecular Clock Activity by Promoting Ubiquitination-Dependent Degradation of the Mammalian CRY1. PLoS ONE 2015, 10, e0139725.
- Tamayo, A.G.; Duong, H.A.; Robles, M.S.; Mann, M.; Weitz, C.J. Histone Monoubiquitination by Clock–Bmal1 Complex Marks Per1 and Per2 Genes for Circadian Feedback. Nat. Struct. Mol. Biol. 2015, 22, 759–766.
- Okazaki, H.; Matsunaga, N.; Fujioka, T.; Okazaki, F.; Akagawa, Y.; Tsurudome, Y.; Ono, M.; Kuwano, M.; Koyanagi, S.; Ohdo, S. Circadian Regulation of mTOR by the Ubiquitin Pathway in Renal Cell Carcinoma. Cancer Res. 2014, 74, 543–551.
- Mao, J.-H.; Kim, I.-J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 Targets mTOR for Degradation and Cooperates with PTEN in Tumor Suppression. Science 2008, 321, 1499–1502.
- Fang, L.; Yang, Z.; Zhou, J.; Tung, J.-Y.; Hsiao, C.-D.; Wang, L.; Deng, Y.; Wang, P.; Wang, J.; Lee, M.-H. Circadian Clock Gene CRY2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1476–1487.
- Zhao, X.; Hirota, T.; Han, X.; Cho, H.; Chong, L.-W.; Lamia, K.; Liu, S.; Atkins, A.R.; Banayo, E.; Liddle, C.; et al. Circadian Amplitude Regulation via FBXW7-Targeted REV-ERBα Degradation. Cell 2016, 165, 1644–1657.
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a Proteome-Scale Map of the Human Protein-Protein Interaction Network. Nature 2005, 437, 1173–1178.
- Chen, S.; Yang, J.; Yang, L.; Zhang, Y.; Zhou, L.; Liu, Q.; Duan, C.; Mieres, C.A.; Zhou, G.; Xu, G. Ubiquitin Ligase TRAF2 Attenuates the Transcriptional Activity of the Core Clock Protein BMAL1 and Affects the Maximal Per1 mRNA Level of the Circadian Clock in Cells. FEBS J. 2018, 285, 2987–3001.
- Ullah, K.; Chen, S.; Lu, J.; Wang, X.; Liu, Q.; Zhang, Y.; Long, Y.; Hu, Z.; Xu, G. The E3 Ubiquitin Ligase STUB1 Attenuates Cell Senescence by Promoting the Ubiquitination and Degradation of the Core Circadian Regulator BMAL1. J. Biol. Chem. 2020, 295, 4696–4708.
- Ehlen, J.C.; Jones, K.A.; Pinckney, L.; Gray, C.L.; Burette, S.; Weinberg, R.J.; Evans, J.A.; Brager, A.J.; Zylka, M.J.; Paul, K.N.; et al. Maternal Ube3a Loss Disrupts Sleep Homeostasis But Leaves Circadian Rhythmicity Largely Intact. J. Neurosci. 2015, 35, 13587–13598.
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP Mutations Cause Angelman Syndrome. Nat. Genet. 1997, 15, 70–73.
- Jones, K.A.; Han, J.E.; DeBruyne, J.P.; Philpot, B.D. Persistent Neuronal Ube3a Expression in the Suprachiasmatic Nucleus of Angelman Syndrome Model Mice. Sci. Rep. 2016, 6, 28238.
- Shi, S.-Q.; Mahoney, C.E.; Houdek, P.; Zhao, W.; Anderson, M.P.; Zhuo, X.; Beaudet, A.; Sumova, A.; Scammell, T.E.; Johnson, C.H. Circadian Rhythms and Sleep Are Dependent Upon Expression Levels of Key Ubiquitin Ligase Ube3a. Front. Behav. Neurosci. 2022, 16, 837523.
- Thibert, R.L.; Larson, A.M.; Hsieh, D.T.; Raby, A.R.; Thiele, E.A. Neurologic Manifestations of Angelman Syndrome. Pediatr. Neurol. 2013, 48, 271–279.
- Colas, D.; Wagstaff, J.; Fort, P.; Salvert, D.; Sarda, N. Sleep Disturbances in Ube3a Maternal-Deficient Mice Modeling Angelman Syndrome. Neurobiol. Dis. 2005, 20, 471–478.
- Shi, S.; Bichell, T.J.; Ihrie, R.A.; Johnson, C.H. Ube3a Imprinting Impairs Circadian Robustness in Angelman Syndrome Models. Curr. Biol. 2015, 25, 537–545.
- Gossan, N.C.; Zhang, F.; Guo, B.; Jin, D.; Yoshitane, H.; Yao, A.; Glossop, N.; Zhang, Y.Q.; Fukada, Y.; Meng, Q.-J. The E3 Ubiquitin Ligase UBE3A Is an Integral Component of the Molecular Circadian Clock through Regulating the BMAL1 Transcription Factor. Nucleic Acids Res. 2014, 42, 5765–5775.
- Ullah, K.; Zubia, E.; Narayan, M.; Yang, J.; Xu, G. Diverse Roles of the E2/E3 Hybrid Enzyme UBE2O in the Regulation of Protein Ubiquitination, Cellular Functions, and Disease Onset. FEBS J. 2019, 286, 2018–2034.
- Chen, S.; Yang, J.; Zhang, Y.; Duan, C.; Liu, Q.; Huang, Z.; Xu, Y.; Zhou, L.; Xu, G. Ubiquitin-Conjugating Enzyme UBE2O Regulates Cellular Clock Function by Promoting the Degradation of the Transcription Factor BMAL1. J. Biol. Chem. 2018, 293, 11296–11309.
- DeBruyne, J.P.; Baggs, J.E.; Sato, T.K.; Hogenesch, J.B. Ubiquitin Ligase Siah2 Regulates RevErbα Degradation and the Mammalian Circadian Clock. Proc. Natl. Acad. Sci. USA 2015, 112, 12420–12425.
- Mekbib, T.; Suen, T.-C.; Rollins-Hairston, A.; Smith, K.; Armstrong, A.; Gray, C.; Owino, S.; Baba, K.; Baggs, J.E.; Ehlen, J.C.; et al. The Ubiquitin Ligase SIAH2 Is a Female-Specific Regulator of Circadian Rhythms and Metabolism. PLoS Genet. 2022, 18, e1010305.
- Chen, D.; Brooks, C.L.; Gu, W. ARF-BP1 as a Potential Therapeutic Target. Br. J. Cancer 2006, 94, 1555–1558.
- Han, S.; Witt, R.M.; Santos, T.M.; Polizzano, C.; Sabatini, B.L.; Ramesh, V. Pam (Protein Associated with Myc) Functions as an E3 Ubiquitin Ligase and Regulates TSC/mTOR Signaling. Cell. Signal. 2008, 20, 1084–1091.
- Yin, L.; Joshi, S.; Wu, N.; Tong, X.; Lazar, M.A. E3 Ligases Arf-Bp1 and Pam Mediate Lithium-Stimulated Degradation of the Circadian Heme Receptor Rev-Erbα. Proc. Natl. Acad. Sci. USA 2010, 107, 11614–11619.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
868
Revisions:
2 times
(View History)
Update Date:
21 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No