Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ioana-Andreea Lungu | -- | 4509 | 2022-09-16 17:58:45 | | | |
| 2 | Conner Chen | + 2 word(s) | 4511 | 2022-09-20 05:57:09 | | | | |
| 3 | Conner Chen | + 10 word(s) | 4521 | 2022-09-22 10:13:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lungu, I. Antibiotic Hybrids. Encyclopedia. Available online: https://encyclopedia.pub/entry/27260 (accessed on 09 August 2026).
Lungu I. Antibiotic Hybrids. Encyclopedia. Available at: https://encyclopedia.pub/entry/27260. Accessed August 09, 2026.
Lungu, Ioana-Andreea. "Antibiotic Hybrids" Encyclopedia, https://encyclopedia.pub/entry/27260 (accessed August 09, 2026).
Lungu, I. (2022, September 16). Antibiotic Hybrids. In Encyclopedia. https://encyclopedia.pub/entry/27260
Lungu, Ioana-Andreea. "Antibiotic Hybrids." Encyclopedia. Web. 16 September, 2022.
Copy Citation
An emerging strategy in the fight against antimicrobial resistance is the development of antibiotic hybrids. The term “hybrid” suggests a two-component molecule with biological activity that retains the activity of the individual components after hybridization, acting synergistically. For example, hybrid drugs that incorporate two active compounds into a single molecule could be used to expand the biological activity and prevent the development of bacterial resistance.
hybrids
antibiotic hybrids
structure–activity relationship
fluoroquinolone hybrids
1. Antibiotic Hybrids as Tools against Antimicrobial Resistance
Spizek and Havlicek (2015) summarized five strategies that could be used to fight the global phenomenon of antibiotic resistance. The first is the development of vaccines that target resistant bacterial strains; secondly, the discovery of new antibiotics (from conventional and less conventional sources); and thirdly, the discovery of new genes that specify the biosynthesis of antibiotics. The fourth strategy is the use and possible adaptation of natural compounds that have fallen out of interest in the present. The last proposed strategy is the discovery of new antibiotic targets [1]. The search for new compounds that possess either natural or synthetic antibiotic effects that are aimed at either traditional or more recent targets still receives interest from scientists [2][3]. Finally, an emerging strategy in the fight against antimicrobial resistance is the development of antibiotic hybrids. Some authors define antibiotic hybrids as “a synthetic construct of two or more pharmacophores belonging to an established agent known to elicit a desired antimicrobial effect” (Domalaon et al., 2018) [4].
The term “hybrid” suggests a two-component molecule with biological activity that retains the activity of the individual components after hybridization, acting synergistically. For example, hybrid drugs that incorporate two active compounds into a single molecule could be used to expand the biological activity and prevent the development of bacterial resistance [5]. Molecular hybridization combines the pharmacophore groups of different bioactive substances to produce a new hybrid molecule with complementary activities and/or multiple pharmacological targets and/or counterbalancing side effects compared to the original molecules. Over the last years, there have been numerous attempts at obtaining and testing these hybrids against various bacterial strains, with many proving successful [5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21].
Quinolones (QNs) and fluoroquinolones (FQNs) are good candidates for hybridization due to their chemical structure, which facilitates linkage with many other active compounds [7]. In addition, other advantages that make FQNs promising for incorporation in antibacterial hybrids are the mechanism of action that confers a bactericidal effect, their effectiveness and potency, and the slower development of antimicrobial resistance [22][23][24].
Prodrug versus Hybrid Comparison
A prodrug is a pharmacologically inactive molecule converted in vivo into active forms by enzymatic or chemical reactions. By designing a prodrug, the pharmacokinetic properties of the active drug (such as bioavailability, absorption, and permeability) can be modified without affecting its pharmacological activity. Prodrugs can be classified into three categories: (1) carrier-linked prodrugs (an active drug linked to a pro-moiety), in which the active drug is released after an enzymatic or chemical reaction by which the moiety is removed; (2) bio-precursor prodrugs (the active drug is modified at the molecular level), where oxidation or reduction reactions modify the structure and release the active drug; and (3) double prodrugs (two biologically active drugs are linked in a single molecule), where the linkers between the two drugs can be cleaved by different mechanisms to release the component molecules [25].
Prodrug design has been used for (F)QNs to improve their physicochemical and pharmacokinetic properties (e.g., water solubility, lipophilicity, absorption, bioavailability) [26][27][28][29]. Some examples of the obtained FQNs’ prodrugs are alatrofloxacin (mesylate salt, a prodrug of trovafloxacin) [30][31][32], bisphosphonated fluoroquinolone esters [30][31][33], polyester prodrugs of norfloxacin [30][31][34], cellulose ether derivatives of ofloxacin [30][31][35], moxifloxacin conjugated with hydrophilic cellulose ethers [30][31][36], alalevonadifloxacin (L-alanine ester prodrug of levonadifloxacin), and N-Acylated ciprofloxacin derivatives [30][31][37][38].
Antibiotic hybrids represent two covalently linked pharmacophores with different mechanisms of action [39]. The design of hybrids (antibiotic–antibiotics or antibiotic–adjuvant) aims to surmount the resistance mechanisms for either or both drugs. The combination with an adjuvant helps by increasing the access to the target site or augmenting the primary antibiotics’ efficacy [30][31].
2. Structural Considerations regarding Antibiotic Hybrids
Although molecules can be directly joined in hybrids, a molecular connector can bind the active molecules together through a covalent bond. The bond can be cleavable or non-cleavable. A hybrid with a cleavable connector would be enzymatically biotransformed when reaching the site of action (the hybrid prodrug approach—mutual prodrug) [25][40]) whilst the non-cleavable linker would remain intact for the duration of its time course in the body (the hybrid drug approach) (Figure 1) [4][30][31]. For example, the valine–citrulline linker is cleavable in the DSTA4637S hybrid [41]. On the other hand, the hybrid named cefiderocol contains a non-cleavable linker [4][42].
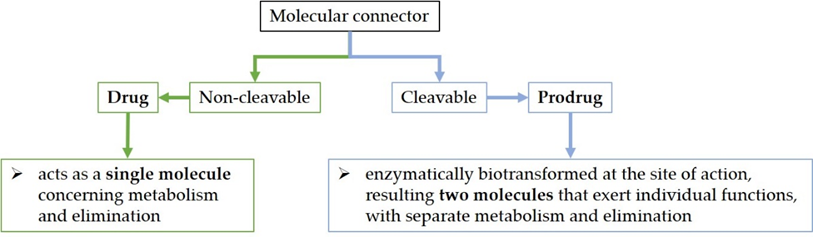
Figure 1. The drug versus prodrug approach.
Regarding an antibiotic hybrid prodrug, the compound is cleaved into two molecules that exert individual functions, with separate metabolism and elimination. On the other hand, the antibiotic hybrid possessing the non-cleavable connector acts as a single molecule concerning metabolism and elimination [30].
Compared to antibiotic combination therapy, antibiotic hybrids would suppress resistance with a single molecular agent, having a single pharmacokinetic profile, while also overcoming the possibility of noncomplementary pharmacodynamics. There is also the premise that hybrid drugs could affect the bacterial strains that are intermediately susceptible or resistant to one of the drug components. Moreover, although uncertain, there is the possibility of retaining antibacterial potency even against pathogens with resistance or intermediate susceptibility to both drug components. Supplementary physicochemical properties lacking in the original molecules could be imparted to the hybrid. It could translate into enhanced efficacy or even a new mechanism of antibacterial action for the obtained hybrid (Figure 2) [4][30].
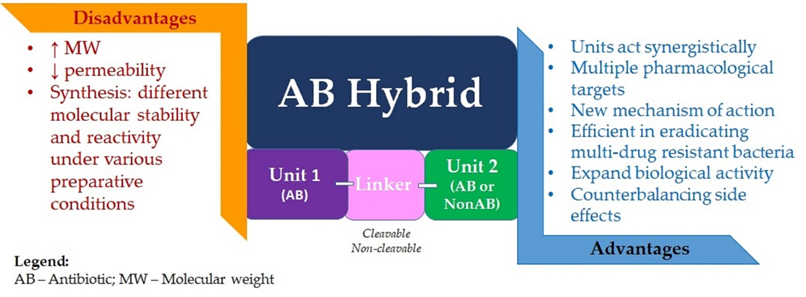
Figure 2. Advantages and disadvantages of antibiotic hybrids.
The concept of an antibiotic hybrid is notably more widespread in the literature than the concept of a prodrug hybrid. An essential challenge in the hybrid prodrug approach is finding a linker specifically cleavable by bacterial enzymes and resistant to human metabolic enzymes. However, both methods require great efforts for synthesis due to the components’ different molecular stability and reactivity under various preparative conditions. Another challenge of designing hybrid drugs is imposed by the characteristic high molecular weight (>600 g/mol) of the resulting molecule; synthesizing agents able to penetrate the dual membrane of Gram-negative bacteria is reasonably difficult. However, several hybrid drugs, efficient in eradicating multi-drug-resistant Gram-negative bacteria and likely capable of delaying drug resistance onset, are currently in preclinical or clinical evaluation, thus bringing hope of a favorable prognosis for this strategy [4][43].
3. Obtained Hybrids with Antibiotics
Examples of antibiotic hybrids in various study phases are presented in Table 1. Many hybrids have been developed to fight Gram-negative bacterial infections [4]. A particular class combines antibiotics with siderophore-type molecules (e.g., cefiderocol) [42]. The siderophores act based on the “Trojan horse” strategy: bacterial iron uptake systems are used, and siderophores enter and destroy bacteria. More macrocycle–antibiotic hybrids are in various stages of development [41].
Table 1. Examples of antibiotic hybrids in various stages of development (AB—antibiotic, LK—linker, NAB—non-antibiotic, C—cleavable, NC—non-cleavable, UTI—urinary tract infection).
| Type | Hybrid (Commercial Name) |
Unit 1 (Class) |
Linker | Unit 2 (Class) |
Possible Indications and Dosage | References |
|---|---|---|---|---|---|---|
| AB-LK-AB | Cadazolid | Tedizolid (oxazolidinones) |
NC | Ciprofloxacin (FQNs) |
Clostridium difficile-associated diarrhea—Phase 1 clinical trial—single oral dose of 3000 mg | [4][44][45] |
| TNP-2092 (CBR-2092) | Rifamycin (ansamycins) |
NC | Ciprofloxacin derivative (FQNs) |
Gastrointestinal and liver disorders—Clostridium difficile infection model—6.67 mg/kg, orally, 7 days, Acute bacterial skin and skin structure infection—Phase 2 clinical trial—300 mg intravenously, every 12 h |
[46][47][48][49] | |
| Cefilavancin (TD-1792) |
Vancomycin (glycopeptide antibiotics) |
NC | THRX-169797 (cephalosporins) | Gram-positive complicated skin and skin structure infections—Phase 2 clinical trial—2 mg/kg/day, intravenously | [41][50][51][52][53] | |
| TD-1607 | Vancomycin (glycopeptide antibiotics) |
C | THRX-169797 (cephalosporins) | Infections with Gram-positive bacteria—Phase 1 clinical trials to evaluate the tolerability, safety, and pharmacokinetics—single escalating doses, intravenously | [41][54] | |
| TNP-2198 | Rifamycin (ansamycins) |
NC | Metronidazole | Helicobacter pylori infection (mouse model), Clostridium difficile infection (hamster model)—5, 15, and 45 mg/kg/day, orally, 5 days;bacterial vaginosis | [41][55] | |
| MCB-3681 | Linezolid (oxazolidinones) |
NC | Ciprofloxacin derivative (FQNs) |
Infections with Gram-positive bacteria—multiple-dose phase 1 study—6 mg/kg body weight over 12 h for 5 days, intravenously | [56] | |
| AB-LK-NAB | Cefiderocol (Fetroja) |
Ceftazidime (cephalosporins) |
NC | 2-chloro-3,4-dihydroxybenzoic acid (catechol derivative; siderophore) |
Complicated UTI and severe carbapenem-resistant Gram-negative bacterial infection—Phase 3 clinical trial—2 g intravenously over 3 h every 8 h for a period of 7 to 14 days, or 2 g every 6 h for participants with creatinine clearance >120 mL/min | [4][57][58][59][60] |
| - | Ampicillin/ Amoxycillin |
NC | Enterobactin (catecholate siderophore) |
Escherichia coli Infections—microbiological assay |
[61] | |
| - | Ampicillin | NC | Tetramic acid(s) | Gram-negative bacterial infections—microbiological assay | [62] | |
| DSTA4637S | 4-Dimethylaminopiperidino-hydroxybenzoxazino rifamycin (ansamycins) |
C | Thiomab human immunoglobulin G1 (IgG1) monoclonal antibody | Staphylococcus aureus infections—Phase 1 clinical trials—low-, intermediate-, and high-dose intravenous infusion | [41][63][64][65][66] |
Additionally, hybrids that include FQNs are numerous and will be presented separately in the following section.
The design of new antibiotics must overcome the passage through the membranes of bacteria. Dual-acting antibiotic hybrids are promising agents to overcome drug resistance in multi-drug-resistant bacteria. However, the high molecular weight (over 600 g/mol) and pharmacokinetic differences of antibiotic hybrids are significant disadvantages for permeability and metabolism [4][67]. On the other hand, it seems that the molecular mass as a criterion for a drug-like compound (Ro5) needs to be updated. Many drugs or prodrugs violate one or even two Ro5 rules (e.g., cyclic peptide immunosuppressants, macrolide antibiotics, HIV protease inhibitors, tyrosine kinase inhibitors, antifungals, anti-cancers). Oral drugs “beyond the Ro5” (bRo5) seem to need a specific degree of flexibility to present aqueous solubility, transport through cell membranes, and target binding [68][69][70]. In 2020, 15 of the 26 drugs approved by the Food and Drug Administration (FDA) (58%) violated one or more drug-likeness pharmacokinetic principles [71]. Therefore, It can be highlighted that antibiotic hybrids cannot be discriminated against based on their high molecular weight without proper fundamental and clinical research. As an alternative, hybrids with antibiotic effects could also be used topically for treating various infections with multi-resistant pathogens. A good example is the hybrid TNP-2198 (Table 1).
Due to technological progress, computer-aided drug design (CADD) methods are beneficial for predicting new molecules with antibacterial activity and designing “hybrid” molecules. Examples of discovered compounds through CADD and bacteria on which they have potential action have been presented by Jukič and Bren (2022) in their review article [72].
4. Hybrids with FQNs
Hybridization of FQNs with other molecules (e.g., aminoglycosides, benzofuroxanes, oxazolidinones, etc.) produces candidates with synergistic antibacterial effect, activity on resistant bacteria, reduced toxicity, or other biological effects. To date, studies have been performed in which FQNs have been included in hybrids with various molecules, both other antibiotics or non-antibiotics (e.g., substances of the aminoglycoside class (ciprofloxacin-neomycin [16], moxifloxacin-tobramycin [11]), oxazolidinones (ciprofloxacin-linezolid [12]), or with benzofuroxan [17] and benzimidazole derivatives [19]) Table 2 comprises the antimicrobial activity of the hybrids presented in the following section.
Table 2. The antimicrobial activity of QN/FQN hybrids (represented through MIC).
| Type of Hybrid | Compound Code | Microorganism | MIC | Reference |
|---|---|---|---|---|
| QN-FQN | 10f | Staphylococcus aureus | 3.3 μM | [73] |
| 10b | Streptococcus pyogenes | 7.8 μM | ||
| 11a | Salmonella typhi | 7.6 μM | ||
| 11b | 7.4 μM | |||
| N-alkylations of the C-7 chain of QN | 7l | Mycobacterium tuberculosis H37Rv and multi-drug-resistant Mycobacterium tuberculosis | 0.09 μM | [74] |
| Oxazolidinone-FQN | 2, 5 and 6 | Staphylococcus aureus Enterococcus faecium |
≤1 μg/mL | [12] |
| Tetracycline-FQN | 10 | Mycobacterium tuberculosis | 0.2 μg/mL | [75] |
| Rifamycin-QN | CBR-2092 | 300 clinical isolates of staphylococci and streptococci | 0.008–0.5 μg/mL | [76] |
| Aminoglycoside-FQN | 1i | Escherichia coli (R477-100, ATCC 25922, AG100B, AG100A) | 0.75–3 μg/mL | [16] |
| 1q | 0.38–12 μg/mL | |||
| Azithromycin-QN | 7f | Streptococcus pyogenes | 0.5 μg/mL | [77] |
| 8f | 1 μg/mL | |||
| 7f | Haemophilus influenzae B 0529 | 0.5 μg/mL | ||
| 8f | 0.5 μg/mL | |||
| Aminoglycoside-FQN | 1 | Staphylococcus aureus and methicillin-resistant Staphylococcus aureus | 1 μg/mL | [11] |
| three Pseudomonas aeruginosa strains (including two gentamicin-resistant Pseudomonas aeruginosa strains) | 4−8 μg/mL | |||
| Aminoglycoside-FQN | 1m | Escherichia coli | 6.2 ± 0.7 μM (day 1) 30.3 ± 3.4 μM (day 17) | [78] |
| Aminoglycoside-FQN | 1b | Escherichia coli (R477-100, 25922, AG100B, AG100A) | 0.37–12 μg/mL | [10] |
| Bacillus subtilis | 1.5 μg/mL | |||
| ATP-competitive inhibitors (for DNA Gyrase A and B)-FQN | 3a | Klebsiella pneumoniae | 0.5 µg/mL | [79] |
| Enterobacter cloacae | 4 µg/mL | |||
| Escherichia coli | 2 µg/mL | |||
| 3-arylfuran-2(5H)-one-FQN | 11 | Multiple drug-resistant Escherichia coli | 0.11 μg/mL | [80] |
| Benzimidazole-QN | 5b | Pseudomonas aeruginosa | 1 μg/mL | [19] |
| Staphylococcus aureus and methicillin-resistant Staphylococcus aureus | 8 μg/mL | |||
| Klebsiella pneumoniae | 16 μg/mL | |||
| Benzofuroxane-FQN | 4d | Bacillus cereus 8035 | 0.97 μg/mL | [17] |
| Flavonoids (naringenin)-FQN | 7 | Escherichia coli | 0.71 μg/mL | [81] |
| Bacillus subtilis | 0.062 μg/mL | |||
| Staphylococcus aureus | 0.29 μg/mL | |||
| Candida albicans | 0.14 μg/mL | |||
| 1,3,4-Oxadiazole-FQN | 4 b–d | Staphylococcus aureus | ≤0.125 μg/mL | [82] |
| Sulfonamide-FQN | 3a | Staphylococcus aureus | 0.324 μM | [83] |
| Escherichia coli ATCC8739 | 0.025 μM | |||
| 3b | Staphylococcus aureus | 0.422 μM | ||
| Escherichia coli ATCC8739 | 0.013 μM | |||
| Triazole-FQN | 11 | Candida albicans | 10.23 µg/mL | [84] |
| Trimethoprim-FQN | BP-4Q-002 | Staphylococcus aureus | 0.5 μg/mL | [85] |
| Escherichia coli | 1 μg/mL | |||
| Staphylococcus aureus NRS19 (resistant to ciprofloxacin) | 1 μg/mL |
4.1. Antibiotic–Antibiotic Hybrids
Most published FQN hybrids present a linker between the two parent molecules. The two antibiotic molecules’ connectors differ from study to study (a carbon unit or more or diverse chemical elements). Figure 3, Figure 4, Figure 5, Figure 6, Figure 7 and Figure 8 illustrate such examples (the linker is highlighted with the orange circle while the blue rectangle highlights the FQN unit) [30][67]. Each research group probably selected the most successful linker and the simplest way to obtain an antibiotic hybrid. Apart from the “cleavable/non-cleavable” classification, the connectors have not yet been classified according to other criteria.
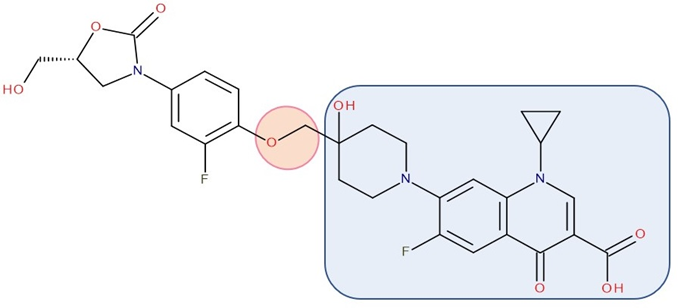
Figure 3. Antibiotic–antibiotic hybrid containing an FQN: tedizolid derivative–linker–ciprofloxacin derivative (Cadazolid); the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [44].
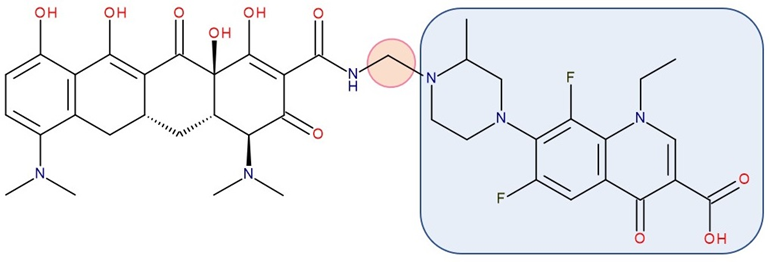
Figure 4. Antibiotic–antibiotic hybrid containing an FQN: minocycline–linker–lomefloxacin; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [75].

Figure 5. Antibiotic–antibiotic hybrid containing an FQN: rifampicin derivative–linker–ciprofloxacin derivative; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [76].
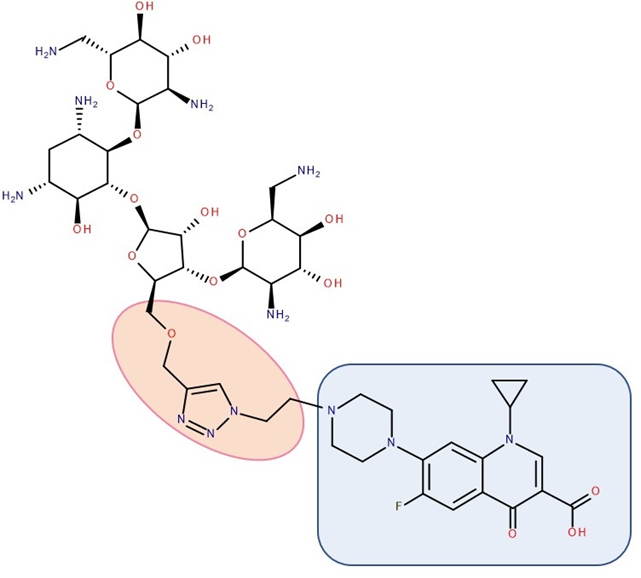
Figure 6. Antibiotic–antibiotic hybrid containing an FQN: neomycin B–linker–ciprofloxacin; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [16].
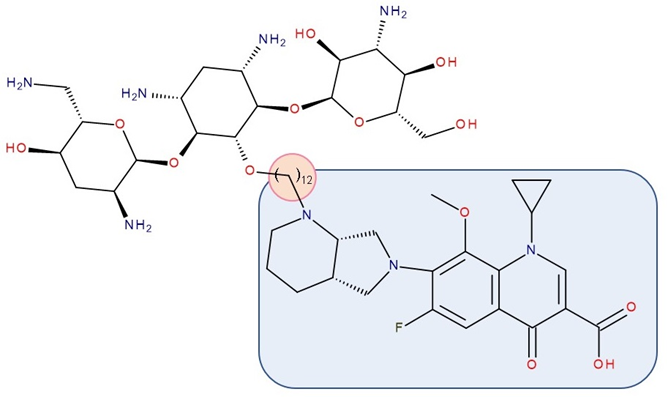
Figure 7. Antibiotic–antibiotic hybrid containing an FQN: tobramycin–linker–moxifloxacin; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [11].
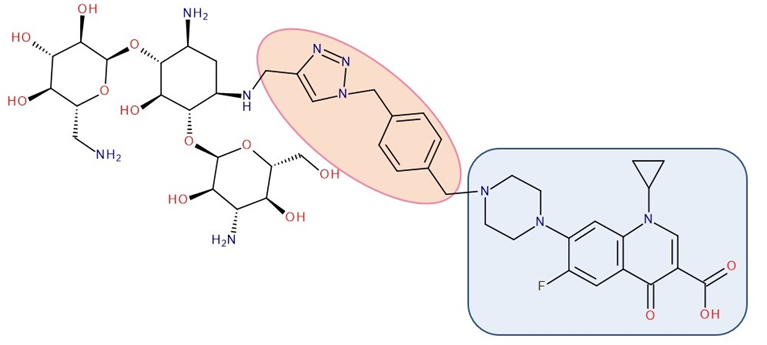
Figure 8. Antibiotic–antibiotic hybrid containing an FQN: kanamycin A–linker–ciprofloxacin; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle [10].
The most advantageous way of binding is to the radical in position 7 of the structure of FQNs, responsible for the antimicrobial potential and pharmacokinetic properties; thus, the groups responsible for binding to the bacterial target enzymes remain unaffected. Among the binding possibilities in the structure of hybrids is the formation of Mannich bases, between tetracyclines, formaldehyde, and the secondary amino group (piperazine) of FQNs [75].
A series of quinolone–fluoroquinolone hybrids were synthesized through benzotriazole chemistry. The C7 positions (the piperazine ring) of ciprofloxacin and norfloxacin and the amino acid linkers were targeted to obtain the final compounds. The obtained hybrids presented antibacterial properties that were comparable with the parent compounds [73].
Various researchers have synthesized ciprofloxacin derivatives using N-alkylations of the C-7 chain to increase the lipophilia and antibacterial potential [74][86]. The combination with oxazolidinone can be achieved by a bridge linking an FQN to the pharmacophore groups of the oxazolidinone derivative [12]. One of the promising hybrids that reached the phase 3 clinical stage is cadazolid. Cadazolid is a hybrid that contains structural elements of an oxazolidinone with an FQN moiety with significant activity against Clostridium difficile (Figure 3) [44][87][88].
Gordeev et al. (2003) synthesized several compounds that incorporated pharmacophore structures of FQNs and oxazolidinones and demonstrated superior potency to linezolid against Gram-positive and Gram-negative bacteria, even for linezolid- and ciprofloxacin-resistant strains of Staphylococcus aureus and Enterococcus faecium. The mechanism of action combined the inhibition of protein synthesis and DNA gyrase and Topoisomerase IV [12].
Representatives from the tetracyclines class (tetracycline, oxytetracycline, and minocycline) were combined with the secondary amino (piperazine) function of FQNs (norfloxacin, lomefloxacin, ciprofloxacin, and gatifloxacin) by Sriram et al. (2007). The results revealed anti-HIV and antitubercular activities, which were most significant for one of the compounds (minocycline-lomefloxacin derived—Figure 4), making it a promising candidate in treating patients with HIV-1 and co-infected with Mycobacterium tuberculosis [75].
CBR-2092 combines rifampicin and QNs in a hybrid antibiotic structure (Figure 5). Studies showed increased bactericidal activity against Staphylococcus aureus exhibited by CBR-2092, superior to that of rifampicin, moxifloxacin, or the combination of rifampicin and moxifloxacin. Furthermore, it is retained against strains that are intermediate or resistant to rifampicin or quinolone. Additionally, this hybrid prevented the development of resistance and was not a substrate for Staphylococcus aureus efflux pumps (NorA or MepA) [76]. Further studies showed that CBR-2092 exhibited a similar potency to rifampicin as an inhibitor of RNA polymerase, inhibited DNA gyrase and DNA Topoisomerase IV, and maintained activity against a variant commonly resistant to quinolone. Furthermore, CBR-2092 showed effects similar to rifampicin on RNA synthesis in strains susceptible to rifampicin and quinolone-like effects on DNA synthesis in strains resistant to rifampicin [89].
In a recent study, kanglemycin A (a rifampicin analogue) was linked to nine FQNs to obtain hybrids with superior antibacterial activity. Kanglemycin presents a dimethyl succinic acid moiety as an offering chemical group in synthesizing antibiotic hybrids. The activity of the synthesized hybrids linked to the acid group versus synthesized hybrids linked at the compound’s naphthoquinone ring system was compared. These have been proven to be determinants of the biological activity of the hybrids [90].
A series of hybrids with ciprofloxacin (FQNs) and neomycin (aminoglycoside) was synthesized by Pokrovskaya et al. (2009) (Figure 6). The antibacterial activity of most of the synthesized compounds was significantly higher than that of neomycin, in particular for Gram-negative bacteria and MRSA. Moreover, they overcame the most common types of aminoglycosides-associated resistance. When treated with the ciprofloxacin–neomycin hybrid, a significant delay in resistance formation against Gram-negative (Escherichia coli) and Gram-positive (Bacillus subtilis) bacteria was observed for the mixture of the two drugs or each drug separately. The hybrids’ mechanism of action could inhibit protein translation similar to or better than neomycin. Most importantly, they inhibited DNA gyrase and Topoisomerase IV up to 32-fold more than ciprofloxacin, proving a dual mechanism of action characteristic of hybrids [16].
Azithromycin and quinolone substructures were conjoined to preserve pharmacophores from both molecules; some obtained representatives showed an improved potency compared to azithromycin against Gram-positive and Gram-negative pathogens. Moreover, they maintained activity against macrolide-lincosamide-streptogramin-resistant strains of Streptococcus pneumoniae and Streptococcus pyogenes. Furthermore, they displayed increased potency over azithromycin and telithromycin against the Gram-negative Haemophilus influenzae [77].
Gorityala et al. (2016) used ciprofloxacin and moxifloxacin to synthesize conjugates with tobramycin (aminoglycoside) (Figure 7). Long carbon chains were used to link the compound molecules. The antibacterial properties were evaluated. In the synthesized series, among some hybrids that exhibited weak antibacterial effects, two of the hybrids showed good antibacterial effects against multi-drug-resistant strains of Pseudomonas aeruginosa. These conjugates destabilized the membrane and inhibited DNA gyrase A and Topoisomerase IV better than the original FQN and reduced efflux. The effect of the aminoglycoside (inhibition of protein translation) was reduced. However, it was observed that the development of bacterial resistance was delayed [11]. The ciprofloxacin–tobramycin hybrid was the first to be electrochemically characterized [91].
The emergence of resistance in Escherichia coli was evaluated for combining ciprofloxacin and neomycin B (aminoglycoside) compared to a hybrid drug obtained from the two antibiotics. The hybrids were synthesized, containing different linkers. For example, an aromatic triazole linker or hydroxyl group-containing aliphatic triazole linker united ciprofloxacin and neomycin B. The authors found that the bacterial populations grown in the presence of the hybrid developed less resistance than those produced in an equimolar mixture of the components. Furthermore, it was found that the ciprofloxacin part of the hybrid was responsible for the inhibition of bacterial growth while the neomycin B part limited resistance mediated by efflux [78]. A series of hybrids composed of ciprofloxacin (FQN) and kanamycin A (aminoglycoside) (Figure 8) were synthesized by Shavit et al. (2017) and showed superior activity against Gram-negative bacteria. These hybrids delayed the emergence of resistance for strains of Escherichia coli and Bacillus subtilis compared to the 1:1 mixture of the two antibiotics [10].
Most of the FQN antibiotic hybrids targeted in the manuscript are studied or are under study regarding their biological effects in vitro [11][12][16][73][74][75][76][77][78][78][86][89][90].
If the antibiotic hybrid contains a cleavable linker, there will be a high chance that the adverse reactions of the hybrid will be those of the FQN unit. However, in the case of a non-cleavable linker, it is possible to reduce the side effects of the FQN unit. Cadazolid is an FQN antibiotic hybrid in clinical phase 3 [44]. Seiler P. et al. (2019) reported that treatment with cadazolid did not lead to one potential side effect, namely the appearance of vancomycin-resistant enterococci, when treating Clostridium difficile infection. Therefore, cadazolid is a promising antibiotic alternative to vancomycin for treating Clostridium difficile infection [92][93]. Currently, few data are published concerning adverse reactions of hybrids with FQNs.
4.2. Antibiotic–Non-Antibiotic Hybrids
Additionally, various hybrids of FQNs with different active substances were synthesized to broaden the antimicrobial spectrum, presented in detail below (Figure 9).
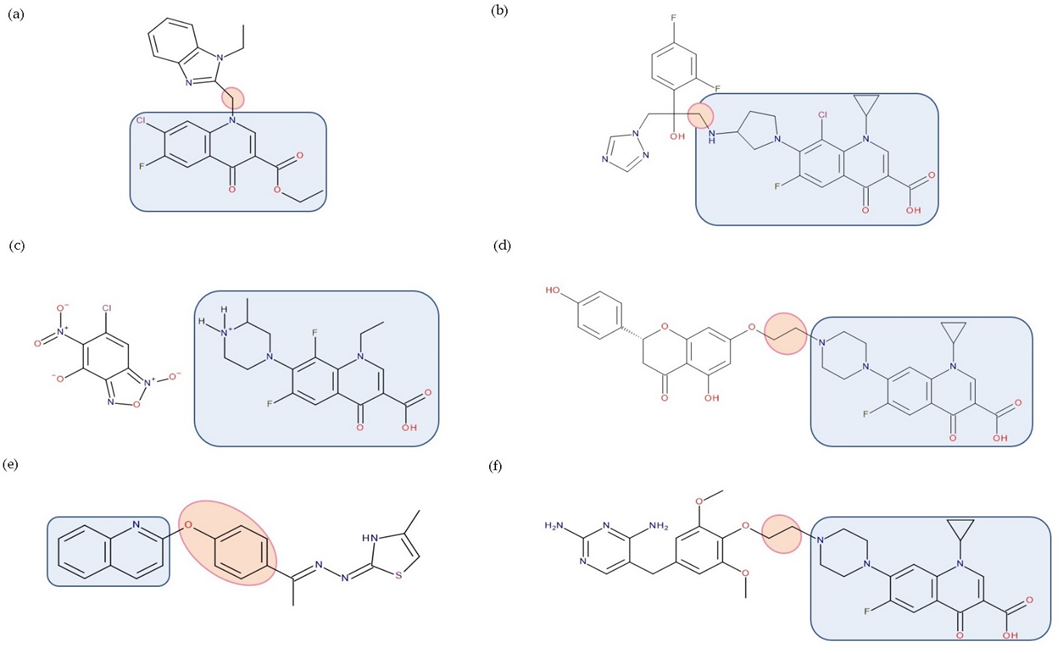
Figure 9. Examples of antibiotic–non-antibiotic hybrids containing an FQN; the linker is highlighted with the orange circle while the QN/FQN unit is highlighted by the blue rectangle: (a) benzimidazole derivative–linker–quinolone derivative (esther form) [19]; (b) triazole derivative–linker–clinafloxacin [94]; (c) benzofuroxan derivative–lomefloxacin [17]; (d) naringenin–linker–ciprofloxacin [4][81]; (e) thiazole derivative–linker–quinolone [95]; (f) trimethoprim–linker–ciprofloxacin [4][85].
Durcik M. et al. (2021) designed and synthesized new hybrids of ciprofloxacin that can interact with the GyrA- and GyrB-binding sites of the target enzyme DNA gyrase. These new compounds demonstrate good activity against Escherichia coli and Klebsiella pneumoniae. In addition, without extensive efflux, some hybrids delayed or prevented the emergence of bacterial resistance [79].
3-Arylfuran-2(5H)-one
An array of covalently linked hybrids between FQNs and a tyrosyl-tRNA synthetase (TyrRS) inhibitor (3-arylfuran-2(5H)-one) was synthesized. Some hybrids displayed activity against both Gram-negative and Gram-positive resistant bacteria. A resulting hybrid of ciprofloxacin exhibited significantly greater potency against MDR Escherichia coli (MIC50—0.11 μg/mL) than the parent FQN (MIC50—5.65 μg/mL, for ciprofloxacin) [80]. This hybrid also displayed a dual mode of action (in vitro), having a more remarkable ability to inhibit DNA gyrase than ciprofloxacin and similar TyrRS inhibitory activity to that of the parent compound [4][80].
Benzimidazole
A series of hybrids between quinolone derivatives and benzimidazole was synthesized by Wang YN et al. (2018). One of the compounds showed remarkable activity against the resistant strains of Pseudomonas aeruginosa and Candida tropicalis. It also caused a decrease in the resistance of Pseudomonas aeruginosa compared to norfloxacin [19].
Benzofuroxane
Chugunova et al. (2016) synthesized a series of FQN hybrids with benzofuroxane derivatives; some hybrids showed superior antibacterial activity on Bacillus cereus 8035 strains compared to the free FQN [17].
Chlorhexidine
Kowalczuk D. et al. (2021) obtained ciprofloxacin–bismuth(III)–chlorhexidine, a new hybrid that contains the bismuth atom as a linker. This new hybrid (metal complex) has potential in the local treatment of wounds. So far, the published data have focused on the structural characteristics of the obtained hybrid using spectroscopic methods [96].
Flavonoids (naringenin)
Another collection of hybrids among FQNs and phenolic flavonoids was obtained. The most compelling representative was between ciprofloxacin and naringenin, with significant activity against methicillin-resistant Staphylococcus aureus (MIC50—0.29 μg/mL), Escherichia coli (MIC50—0.71 μg/mL), and amphotericin B-resistant Candida albicans (MIC50—0.14 μg/mL) [81]. This example also backs up the dual mode of action theory, displaying significant inhibition of both the DNA gyrase (specific to ciprofloxacin) and efflux pump (specific to naringenin) [81][97].
1,3,4-Oxadiazole
In a recent study, 1,3,4-oxadiazole derivatives were linked to the piperazine ring of ciprofloxacin or norfloxacin. Hybrids of the two FQNs with activity against Gram-positive bacteria were obtained. The molecular docking study revealed a high binding affinity for the hybrids 4 c for Topoisomerase IV with a minimum binding energy [82].
Sulfonamides
Nineteen novel ciprofloxacin-sulfonamide hybrid molecules showed significant antibacterial activity. In addition to biological activity, the side effects of hybrids were also tested. The following were used as linkers: azide, acetamide, propionamide, and isopropionamide. The most active hybrids presented lower CNS adverse reactions and GABA expression compared to those that used FQN [83].
Thiazole
The thiazole structural fragment is known for its numerous biological effects in medicinal chemistry research. A series of two-substituted quinolines, including a thiazole moiety separated by a hydrophobic linker, were synthesized and tested against Gram-positive and Gram-negative bacteria. The best structural element for antibacterial activity was the 2,3-dihydrothiazole fragment near the electron-donating group on the nitrogen atom of thiazole and the methyl at the carbon of azomethine. The authors of this study used the model of FQN hybrids with other antibiotics or sulfonamides, keeping the quinoline nucleus in the newly synthesized hybrids [95].
Triazole
An array of clinafloxacin triazole hybrids was synthesized, and their antimicrobial and antifungal activity were evaluated. Most compounds showed similar or better activity against the tested strains (Gram-positive bacteria—four strains, Gram-negative bacteria—four strains, and fungi—two strains) compared to chloramphenicol, clinafloxacin, and fluconazole. Moreover, clinafloxacin triazoles displayed improved efficacy on methicillin-resistant Staphylococcus aureus than clinafloxacin [94].
Another example of FQNs–triazole derivatives is provided by the study performed by Ezelarab et al. (2018). The antifungal activity of a ciprofloxacin–azole hybrid was evaluated, revealing promising results (MIC 10.23 µg/mL, comparable to itraconazole 11.22 µg/mL). Moreover, this obtained hybrid can reasonably bind to the active site of the target (lanosterol 14-α-demethylase CYP51) [84]. Other hybrids with FQNs were synthesized (1,2,3-triazole-substituted ciprofloxacin and norfloxacin derivatives); antibacterial and antifungal activities were investigated in silico and in vitro [98].
The use of the quinolone nucleus in hybrid compounds to obtain antimicrobial activity is supported by a recent study in which hybrids with quinolone derivatives and triazole were obtained. Triazole-linked quinoline derivatives from 8-aminoquinoline presented promising activities against Gram-positive and Gram-negative bacteria and some fungi strains [99]. Other 1H-1,2,3-triazole-linked quinoline–isatin hybrids were recently synthesized by Awolade P. et al. (2021); these new hybrids are promising anti-breast cancer and anti-MRSA agents [100].
N-substituted trifluoroacetimidoyl chlorides
Darehkordi et al. (2011) used N-substituted trifluoroacetimidoyl chlorides to synthesize piperazinyl-quinolone derivatives. Out of the obtained compounds, two exhibited superior antibacterial activity against strains of Escherichia coli, Klebsiella pneumoniae (compared to ciprofloxacin), and Staphylococcus aureus (compared to vancomycin) [9].
Trimethoprim
Although trimethoprim is not used in single therapy as an antibiotic, it was targeted by the hybridization strategy with an FQN. Trimethoprim linked to ciprofloxacin (through the piperazine ring) yielded a hybrid (BP-4Q-002) with good activity against Staphylococcus aureus (MIC 0.5 μg/mL) and Escherichia coli (MIC 1 μg/mL). Against the Staphylococcus aureus strain NRS19 (resistant to ciprofloxacin (MIC for ciprofloxacin—32 μg/mL, minimum inhibitory concentration (MIC) for trimethoprim—4 μg/mL, and MIC for the equimolar mixture—8 μg/mL)), this hybrid exhibited an MIC value of 1 μg/mL) [85]. The activity of BP-4Q-002 against the drug-resistant Staphylococcus aureus strain endorses the concept that hybrid drugs may be able to eradicate strains resistant or intermediately susceptible to one of the parent compounds. Another contribution to a fundamental hypothesis of hybrid drugs (claiming that further qualities are imparted to the hybrid, which is missing in the parent components or the equimolar mixture) could be highlighted by the reduction in the MIC in the case of BP-4Q-002 against the Staphylococcus aureus strain NRS19, compared to the MIC of just ciprofloxacin, trimethoprim, or an equimolar mixture of the two [4].
5. FQN Hybrids with Other Biological Effects
FQNs are being studied for numerous other biological effects [101].
Thus, in addition to the hybrids’ antibacterial properties, as highlighted above for the antifungal effect [19][81][84], quinolone hybrid compounds also showed anti-HIV [75], antifungal [84][94][98], antiplasmodic/antimalarial [102][103], and antitumor [104] potential.
A considerable number of quinolone-based derivatives were synthesized for their antiplasmodial activity to be evaluated. Some displayed promising antiplasmodial (in vitro) activity against chloroquine-sensitive, chloroquine-resistant, and multi-drug-resistant strains of Plasmodium falciparum. At the same time, some showed significant antiplasmodial (in vitro) and antimalarial (in vivo) activity [102][103].
N-4-piperazinyl–ciprofloxacin chalcone hybrids were synthesized, and their activity against various cancer cell lines and topoisomerase inhibitory activity were evaluated. The obtained hybrids exhibited significant inhibitory activity on Topoisomerase I and II while a few of the compounds displayed broad antitumor activity [104].
Additionally, nitric oxide (NO) photo-donor of ciprofloxacin and norfloxacin hybrids were synthesized by Fallica, A.N. et al. (2021) to study the potential anticancer effect. This study showed that some hybrids have intense antiproliferative activity on breast cancer cell lines (aggressive, refractory, and multi-drug-resistant cancer type) [105].
References
- Spizek, J.; Havlicek, V. Tackling Antibiotic Resistance. In Antibiotics: Current Innovations and Future Trends; Caister Academic Press: Wymondham, UK, 2015; pp. 83–94. ISBN 978-1-908230-54-6.
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed Engl. 2016, 55, 6600–6626.
- Fisher, J.F.; Mobashery, S. Endless Resistance. Endless Antibiotics? MedChemComm 2016, 7, 37–49.
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic Hybrids: The Next Generation of Agents and Adjuvants against Gram-Negative Pathogens? Clin. Microbiol. Rev. 2018, 31, e00077-17.
- Gao, F.; Wang, P.; Yang, H.; Miao, Q.; Ma, L.; Lu, G. Recent Developments of Quinolone-Based Derivatives and Their Activities against Escherichia Coli. Eur. J. Med. Chem. 2018, 157, 1223–1248.
- Zhang, G.-F.; Zhang, S.; Pan, B.; Liu, X.; Feng, L.-S. 4-Quinolone Derivatives and Their Activities against Gram Positive Pathogens. Eur. J. Med. Chem. 2018, 143, 710–723.
- Fedorowicz, J.; Sączewski, J. Modifications of Quinolones and Fluoroquinolones: Hybrid Compounds and Dual-Action Molecules. Mon. Chem. 2018, 149, 1199–1245.
- Gao, C.; Fan, Y.-L.; Zhao, F.; Ren, Q.-C.; Wu, X.; Chang, L.; Gao, F. Quinolone Derivatives and Their Activities against Methicillin-Resistant Staphylococcus Aureus (MRSA). Eur. J. Med. Chem. 2018, 157, 1081–1095.
- Darehkordi, A.; Javanmiri, M.; Ghazi, S.; Assar, S. Synthesis of N-Aryl-2,2,2-Trifluoroacetimidoyl Piperazinylquinolone Derivatives and Their Antibacterial Evaluations. J. Fluor. Chem. 2011, 132, 263–268.
- Shavit, M.; Pokrovskaya, V.; Belakhov, V.; Baasov, T. Covalently Linked Kanamycin–Ciprofloxacin Hybrid Antibiotics as a Tool to Fight Bacterial Resistance. Small Mol. Enabled Chem. Biol. Drug Discov. 2017, 25, 2917–2925.
- Gorityala, B.K.; Guchhait, G.; Goswami, S.; Fernando, D.M.; Kumar, A.; Zhanel, G.G.; Schweizer, F. Hybrid Antibiotic Overcomes Resistance in P. Aeruginosa by Enhancing Outer Membrane Penetration and Reducing Efflux. J. Med. Chem. 2016, 59, 8441–8455.
- Gordeev, M.F.; Hackbarth, C.; Barbachyn, M.R.; Banitt, L.S.; Gage, J.R.; Luehr, G.W.; Gomez, M.; Trias, J.; Morin, S.E.; Zurenko, G.E.; et al. Novel Oxazolidinone-Quinolone Hybrid Antimicrobials. Bioorg. Med. Chem. 2003, 13, 4213–4216.
- Marc, G.; Araniciu, C.; Oniga, S.D.; Vlase, L.; Pîrnău, A.; Nadăș, G.C.; Novac, C.Ș.; Matei, I.A.; Chifiriuc, M.C.; Măruțescu, L.; et al. Design, Synthesis and Biological Evaluation of New Piperazin-4-Yl-(Acetyl-Thiazolidine-2,4-Dione) Norfloxacin Analogues as Antimicrobial Agents. Molecules 2019, 24, 3959.
- Liu, L.; Shao, L.; Li, J.; Cui, H.; Li, B.; Zhou, X.; Lv, P.; Zhang, J. Synthesis, Antibacterial Activities, Mode of Action and Acute Toxicity Studies of New Oxazolidinone-Fluoroquinolone Hybrids. Mol. Basel Switz. 2019, 24, 1641.
- Akhtar, R.; Yousaf, M.; Naqvi, S.A.R.; Irfan, M.; Zahoor, A.F.; Hussain, A.I.; Chatha, S.A.S. Synthesis of Ciprofloxacin-Based Compounds: A Review. Synth. Commun. 2016, 46, 1849–1879.
- Pokrovskaya, V.; Belakhov, V.; Hainrichson, M.; Yaron, S.; Baasov, T. Design, Synthesis, and Evaluation of Novel Fluoroquinolone−Aminoglycoside Hybrid Antibiotics. J. Med. Chem. 2009, 52, 2243–2254.
- Chugunova, E.; Akylbekov, N.; Bulatova, A.; Gavrilov, N.; Voloshina, A.; Kulik, N.; Zobov, V.; Dobrynin, A.; Syakaev, V.; Burilov, A. Synthesis and Biological Evaluation of Novel Structural Hybrids of Benzofuroxan Derivatives and Fluoroquinolones. Eur. J. Med. Chem. 2016, 116, 165–172.
- Hu, Y.-Q.; Zhang, S.; Xu, Z.; Lv, Z.-S.; Liu, M.-L.; Feng, L.-S. 4-Quinolone Hybrids and Their Antibacterial Activities. Eur. J. Med. Chem. 2017, 141, 335–345.
- Wang, Y.-N.; Bheemanaboina, R.R.Y.; Gao, W.-W.; Kang, J.; Cai, G.-X.; Zhou, C.-H. Discovery of Benzimidazole-Quinolone Hybrids as New Cleaving Agents toward Drug-Resistant Pseudomonas Aeruginosa DNA. ChemMedChem 2018, 13, 1004–1017.
- Xu, J.-H.; Fan, Y.-L.; Zhou, J. Quinolone–Triazole Hybrids and Their Biological Activities. J. Heterocycl. Chem. 2018, 55, 1854–1862.
- Ji, C.; Xu, X. Recent Advancements in Macrolide Hybrids against Staphylococcus Aureus. Curr. Top. Med. Chem. 2021, 21, 2455–2473.
- Emmerson, A.M.; Jones, A.M. The Quinolones: Decades of Development and Use. J. Antimicrob. Chemother. 2003, 51 (Suppl. 1), 13–20.
- Zhanel, G.G.; Walkty, A.; Vercaigne, L.; Karlowsky, J.A.; Embil, J.; Gin, A.S.; Hoban, D.J. The New Fluoroquinolones: A Critical Review. Can. J. Infect. Dis. 1999, 10, 207–238.
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J.V. Fluoroquinolone Resistance: Mechanisms, Impact on Bacteria, and Role in Evolutionary Success. Trends Microbiol. 2014, 22, 438–445.
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Mol. Basel Switz. 2020, 25, 1543.
- Baker, W.R.; Cai, S.; Dimitroff, M.; Fang, L.; Huh, K.K.; Ryckman, D.R.; Shang, X.; Shawar, R.M.; Therrien, J.H. A Prodrug Approach toward the Development of Water Soluble Fluoroquinolones and Structure—Activity Relationships of Quinoline-3-Carboxylic Acids. J. Med. Chem. 2004, 47, 4693–4709.
- Sharma, P.C.; Piplani, M.; Mittal, M.; Pahwa, R. Insight into Prodrugs of Quinolones and Fluoroquinolones. Infect. Disord. Drug Targets 2016, 16, 140–161.
- Piplani, M.; Rajak, H.; Sharma, P.C. Synthesis and Characterization of N-Mannich Based Prodrugs of Ciprofloxacin and Norfloxacin: In Vitro Anthelmintic and Cytotoxic Evaluation. J. Adv. Res. 2017, 8, 463–470.
- Sharma, P.C.; Piplani, M.; Rajak, H. Synthesis, Characterization and Antimicrobial Evaluation of Lipid Based Norfloxacin Prodrug. Curr. Drug Deliv. 2018, 15, 219–226.
- Gupta, V.; Datta, P. Next-Generation Strategy for Treating Drug Resistant Bacteria: Antibiotic Hybrids. Indian J. Med. Res. 2019, 149, 97–106.
- Pokrovskaya, V.; Baasov, T. Dual-Acting Hybrid Antibiotics: A Promising Strategy to Combat Bacterial Resistance. Expert Opin. Drug Discov. 2010, 5, 883–902.
- Hall, I.H.; Schwab, U.E.; Ward, E.S.; Ives, T.J. Effects of Alatrofloxacin, the Parental Prodrug of Trovafloxacin, on Phagocytic, Anti-Inflammatory and Immunomodulation Events of Human THP-1 Monocytes. Biomed. Pharmacother. 2003, 57, 359–365.
- Tanaka, K.S.E.; Houghton, T.J.; Kang, T.; Dietrich, E.; Delorme, D.; Ferreira, S.S.; Caron, L.; Viens, F.; Arhin, F.F.; Sarmiento, I.; et al. Bisphosphonated Fluoroquinolone Esters as Osteotropic Prodrugs for the Prevention of Osteomyelitis. Bioorg. Med. Chem. 2008, 16, 9217–9229.
- Sobczak, M.; Witkowska, E.; Oledzka, E.; Kolodziejski, W. Synthesis and Structural Analysis of Polyester Prodrugs of Norfloxacin. Molecules 2008, 13, 96–106.
- Amin, M.; Abbas, N.; Hussain, M.; Edgar, K.; Tahir, M.; Tremel, W.; Sher, M. Cellulose Ether Derivatives: A New Platform for Prodrug Formation of Fluoroquinolone Antibiotics. Cellulose 2015, 22, 2011–2022.
- Abbas, N.S.; Amin, M.; Hussain, M.A.; Edgar, K.J.; Tahir, M.N.; Tremel, W. Extended Release and Enhanced Bioavailability of Moxifloxacin Conjugated with Hydrophilic Cellulose Ethers. Carbohydr. Polym. 2016, 136, 1297–1306.
- Bhawsar, S.; Kale, R.; Deshpande, P.; Yeole, R.; Bhagwat, S.; Patel, M. Design and Synthesis of an Oral Prodrug Alalevonadifloxacin for the Treatment of MRSA Infection. Bioorg. Med. Chem. Lett. 2021, 54, 128432.
- Rodvold, K.A.; Gotfried, M.H.; Chugh, R.; Gupta, M.; Yeole, R.; Patel, A.; Bhatia, A. Intrapulmonary Pharmacokinetics of Levonadifloxacin Following Oral Administration of Alalevonadifloxacin to Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62, e02297-17.
- Bremner, J.B.; Ambrus, J.I.; Samosorn, S. Dual Action-Based Approaches to Antibacterial Agents. Curr. Med. Chem. 2007, 14, 1459–1477.
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of Terms Used in Medicinal Chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143.
- Surur, A.S.; Sun, D. Macrocycle-Antibiotic Hybrids: A Path to Clinical Candidates. Front. Chem. 2021, 9, 659845.
- El-Lababidi, R.M.; Rizk, J.G. Cefiderocol: A Siderophore Cephalosporin. Ann. Pharmacother. 2020, 54, 1215–1231.
- Antibiotics Currently in Global Clinical Development. Available online: http://pew.org/1YkUFkT (accessed on 11 June 2022).
- Gerding, D.N.; Cornely, O.A.; Grill, S.; Kracker, H.; Marrast, A.C.; Nord, C.E.; Talbot, G.H.; Buitrago, M.; Diaconescu, I.G.; de Oliveira, C.M.; et al. Cadazolid for the Treatment of Clostridium Difficile Infection: Results of Two Double-Blind, Placebo-Controlled, Non-Inferiority, Randomised Phase 3 Trials. Lancet Infect. Dis. 2019, 19, 265–274.
- Actelion. A Phase 1, Open-Label, Single Oral Dose Study to Investigate the Pharmacokinetics, Safety, and Tolerability of Cadazolid in Patients With Severe Clostridium Difficile Infection (CDI); Actelion: Allschwil, Switzerland, 2014.
- Ma, Z.; Lynch, A.S. Development of a Dual-Acting Antibacterial Agent (TNP-2092) for the Treatment of Persistent Bacterial Infections. J. Med. Chem. 2016, 59, 6645–6657.
- Yuan, Y.; Wang, X.; Xu, X.; Liu, Y.; Li, C.; Yang, M.; Yang, Y.; Ma, Z. Evaluation of a Dual-Acting Antibacterial Agent, TNP-2092, on Gut Microbiota and Potential Application in the Treatment of Gastrointestinal and Liver Disorders. ACS Infect. Dis. 2020, 6, 820–831.
- TenNor Therapeutics Limited. Phase 2, Double-Blind, Randomized, Multicenter, Parallel, Controlled Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of TNP-2092 to Treat Acute Bacterial Skin and Skin Structure Infection in Adults; TenNor Therapeutics Limited: Westfield, NJ, USA, 2020.
- Adams, R.A.; Leon, G.; Miller, N.M.; Reyes, S.P.; Thantrong, C.H.; Thokkadam, A.M.; Lemma, A.S.; Sivaloganathan, D.M.; Wan, X.; Brynildsen, M.P. Rifamycin Antibiotics and the Mechanisms of Their Failure. J. Antibiot. (Tokyo) 2021, 74, 786–798.
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. ACS Infect. Dis. 2018, 4, 715–735.
- Theravance Biopharma. A Phase 2, Randomized, Double Blind, Study of Intravenous TD 1792 Versus Vancomycin for Treatment of Complicated Gram Positive Skin and Skin Structure Infections; Theravance Biopharma: San Francisco, CA, USA, 2021.
- Stryjewski, M.E.; Potgieter, P.D.; Li, Y.-P.; Barriere, S.L.; Churukian, A.; Kingsley, J.; Corey, G.R. TD-1792 versus Vancomycin for Treatment of Complicated Skin and Skin Structure Infections. Antimicrob. Agents Chemother. 2012, 56, 5476–5483.
- Bifunctional Beta-Lactam Antibiotics. Available online: https://encyclopedia.pub/entry/7047 (accessed on 27 July 2022).
- Theravance Biopharma. A Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose (SAD) Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of TD-1607, a Glycopeptide-Cephalosporin Heterodimer Gram-Positive Antibiotic, in Healthy Subjects; Theravance Biopharma: San Francisco, CA, USA, 2021.
- Ma, Z.; He, S.; Yuan, Y.; Zhuang, Z.; Liu, Y.; Wang, H.; Chen, J.; Xu, X.; Ding, C.; Molodtsov, V.; et al. Design, Synthesis, and Characterization of TNP-2198, a Dual-Targeted Rifamycin-Nitroimidazole Conjugate with Potent Activity against Microaerophilic and Anaerobic Bacterial Pathogens. J. Med. Chem. 2022, 65, 4481–4495.
- Dalhoff, A.; Rashid, M.-U.; Kapsner, T.; Panagiotidis, G.; Weintraub, A.; Nord, C.E. Analysis of Effects of MCB3681, the Antibacterially Active Substance of Prodrug MCB3837, on Human Resident Microflora as Proof of Principle. Clin. Microbiol. Infect. 2015, 21, 767.e1–767.e4.
- Commissioner, O. FDA Approves New Antibacterial Drug to Treat Complicated Urinary Tract Infections as Part of Ongoing Efforts to Address Antimicrobial Resistance. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug-treat-complicated-urinary-tract-infections-part-ongoing-efforts (accessed on 26 July 2022).
- Wu, J.Y.; Srinivas, P.; Pogue, J.M. Cefiderocol: A Novel Agent for the Management of Multidrug-Resistant Gram-Negative Organisms. Infect. Dis. Ther. 2020, 9, 17–40.
- Yao, J.; Wang, J.; Chen, M.; Cai, Y. Cefiderocol: An Overview of Its in-Vitro and in-Vivo Activity and Underlying Resistant Mechanisms. Front. Med. 2021, 8, 741940.
- Shionogi. A Multicenter, Randomized, Open-Label Clinical Study of S-649266 or Best Available Therapy for the Treatment of Severe Infections Caused by Carbapenem-Resistant Gram-Negative Pathogens; Shionogi: Osaka, Japan, 2020.
- Zheng, T.; Nolan, E.M. Enterobactin-Mediated Delivery of β-Lactam Antibiotics Enhances Antibacterial Activity against Pathogenic Escherichia Coli. J. Am. Chem. Soc. 2014, 136, 9677–9691.
- Cherian, P.T.; Deshpande, A.; Cheramie, M.N.; Bruhn, D.F.; Hurdle, J.G.; Lee, R.E. Design, Synthesis and Microbiological Evaluation of Ampicillin-Tetramic Acid Hybrid Antibiotics. J. Antibiot. (Tokyo) 2017, 70, 65–72.
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L.; et al. A Phase 1, Randomized, Single-Ascending-Dose Study To Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus Aureus Thiomab Antibody-Antibiotic Conjugate, in Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63, e02588-18.
- Vogt, J.; Hazenbos, W.; Pillow, T.; Proctor, W. P49-Uncovering Novel Lysosomal PH Changes Caused by DmDNA31, a Novel Rifalog Payload of an Antibody-Antibiotic Conjugate (AAC) in Development to Treat Staphylococcus Aureus Infections. Drug Metab. Pharmacokinet. 2020, 35, S36.
- Genentech, Inc. A Phase IB, Randomized, Double-Blind, Placebo-Controlled, Multiple-Ascending Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S in Patients With Staphylococcus Aureus Bacteremia Receiving Standard-of-Care Antibiotics; Genentech, Inc.: San Francisco, CA, USA, 2020.
- Genentech, Inc. A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single-Ascending Dose Study To Investigate The Safety, Tolerability, And Pharmacokinetics Of Dsta4637s In Healthy Volunteers; Genentech, Inc.: San Francisco, CA, USA, 2018.
- Parkes, A.L.; Yule, I.A. Hybrid Antibiotics-Clinical Progress and Novel Designs. Expert Opin. Drug Discov. 2016, 11, 665–680.
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98.
- Doak, B.C.; Kihlberg, J. Drug Discovery beyond the Rule of 5-Opportunities and Challenges. Expert Opin. Drug Discov. 2017, 12, 115–119.
- Protti, Í.F.; Rodrigues, D.R.; Fonseca, S.K.; Alves, R.J.; de Oliveira, R.B.; Maltarollo, V.G. Do Drug-Likeness Rules Apply to Oral Prodrugs? ChemMedChem 2021, 16, 1446–1456.
- Pathania, S.; Singh, P.K. Analyzing FDA-Approved Drugs for Compliance of Pharmacokinetic Principles: Should There Be a Critical Screening Parameter in Drug Designing Protocols? Expert Opin. Drug Metab. Toxicol. 2021, 17, 351–354.
- Jukič, M.; Bren, U. Machine Learning in Antibacterial Drug Design. Front. Pharmacol. 2022, 13, 864412.
- Panda, S.S.; Liaqat, S.; Girgis, A.S.; Samir, A.; Hall, C.D.; Katritzky, A.R. Novel Antibacterial Active Quinolone-Fluoroquinolone Conjugates and 2D-QSAR Studies. Bioorg. Med. Chem. Lett. 2015, 25, 3816–3821.
- Senthilkumar, P.; Dinakaran, M.; Yogeeswari, P.; China, A.; Nagaraja, V.; Sriram, D. Antimycobacterial Activities of Novel Fluoroquinolones. Biomed. Pharmacother. 2009, 63, 27–35.
- Sriram, D.; Yogeeswari, P.; Senchani, G.; Banerjee, D. Newer Tetracycline Derivatives: Synthesis, Anti-HIV, Antimycobacterial Activities and Inhibition of HIV-1 Integrase. Bioorg. Med. Chem. Lett. 2007, 17, 2372–2375.
- Robertson, G.T.; Bonventre, E.J.; Doyle, T.B.; Du, Q.; Duncan, L.; Morris, T.W.; Roche, E.D.; Yan, D.; Lynch, A.S. In Vitro Evaluation of CBR-2092, a Novel Rifamycin-Quinolone Hybrid Antibiotic: Microbiology Profiling Studies with Staphylococci and Streptococci. Antimicrob. Agents Chemother. 2008, 52, 2324–2334.
- Pavlović, D.; Mutak, S. Discovery of 4′′-Ether Linked Azithromycin-Quinolone Hybrid Series: Influence of the Central Linker on the Antibacterial Activity. ACS Med. Chem. Lett. 2011, 2, 331–336.
- Wang, K.K.; Stone, L.K.; Lieberman, T.D.; Shavit, M.; Baasov, T.; Kishony, R. A Hybrid Drug Limits Resistance by Evading the Action of the Multiple Antibiotic Resistance Pathway. Mol. Biol. Evol. 2016, 33, 492–500.
- Durcik, M.; Skok, Ž.; Ilaš, J.; Zidar, N.; Zega, A.; Szili, P.É.; Draskovits, G.; Révész, T.; Kikelj, D.; Nyerges, A.; et al. Hybrid Inhibitors of DNA Gyrase A and B: Design, Synthesis and Evaluation. Pharmaceutics 2021, 13, 6.
- Wang, X.-D.; Wei, W.; Wang, P.-F.; Tang, Y.-T.; Deng, R.-C.; Li, B.; Zhou, S.-S.; Zhang, J.-W.; Zhang, L.; Xiao, Z.-P.; et al. Novel 3-Arylfuran-2(5H)-One-Fluoroquinolone Hybrid: Design, Synthesis and Evaluation as Antibacterial Agent. Bioorg. Med. Chem. 2014, 22, 3620–3628.
- Xiao, Z.-P.; Wang, X.-D.; Wang, P.-F.; Zhou, Y.; Zhang, J.-W.; Zhang, L.; Zhou, J.; Zhou, S.-S.; Ouyang, H.; Lin, X.-Y.; et al. Design, Synthesis, and Evaluation of Novel Fluoroquinolone-Flavonoid Hybrids as Potent Antibiotics against Drug-Resistant Microorganisms. Eur. J. Med. Chem. 2014, 80, 92–100.
- Yang, P.; Luo, J.-B.; Zhang, L.-L.; Wang, Y.-S.; Xie, X.-B.; Shi, Q.-S.; Zhang, X.-G. Design, Synthesis and Antibacterial Studies of 1,3,4-Oxadiazole-Fluoroquinolone Hybrids and Their Molecular Docking Studies. ChemistrySelect 2021, 6, 13209–13214.
- Ibrahim, N.M.; Fahim, S.H.; Hassan, M.; Farag, A.E.; Georgey, H.H. Design and Synthesis of Ciprofloxacin-Sulfonamide Hybrids to Manipulate Ciprofloxacin Pharmacological Qualities: Potency and Side Effects. Eur. J. Med. Chem. 2022, 228, 114021.
- Ezelarab, H.A.A.; Hassan, H.A.; Abbas, S.H.; Abd El-Baky, R.M.; Aburahama, G.E.-D.A. Design, Synthesis and Antifungal Activity of 1,2,4-Triazole/ or 1,3,4-Oxadiazole-Ciprofloxacin Hybrids. J. Adv. Biomed. Pharm. Sci. 2018, 1, 78–84.
- Labischinski, H.; Cherian, J.; Calanasan, C.; Boyce, R.S. Hybrid Antimicrobial Compounds and Their. Use. Patent WO2010025906A2, 11 March 2010.
- Hagihara, M.; Kashiwase, H.; Katsube, T.; Kimura, T.; Komai, T.; Momota, K.; Ohmine, T.; Nishigaki, T.; Kimura, S.; Shimada, K. Synthesis and Anti-HIV Activity of Arylpiperazinyl Fluoroquinolones: A New Class of Anti-HIV Agents. Bioorg. Med. Chem. Lett. 1999, 9, 3063–3068.
- Endres, B.T.; Bassères, E.; Alam, M.J.; Garey, K.W. Cadazolid for the Treatment of Clostridium Difficile. Expert Opin. Investig. Drugs 2017, 26, 509–514.
- Scaiola, A.; Leibundgut, M.; Boehringer, D.; Caspers, P.; Bur, D.; Locher, H.H.; Rueedi, G.; Ritz, D. Structural Basis of Translation Inhibition by Cadazolid, a Novel Quinoxolidinone Antibiotic. Sci. Rep. 2019, 9, 5634.
- Robertson, G.T.; Bonventre, E.J.; Doyle, T.B.; Du, Q.; Duncan, L.; Morris, T.W.; Roche, E.D.; Yan, D.; Lynch, A.S. In Vitro Evaluation of CBR-2092, a Novel Rifamycin-Quinolone Hybrid Antibiotic: Studies of the Mode of Action in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2008, 52, 2313–2323.
- Peek, J.; Koirala, B.; Brady, S.F. Synthesis and Evaluation of Dual-Action Kanglemycin-Fluoroquinolone Hybrid Antibiotics. Bioorg. Med. Chem. Lett. 2022, 57, 128484.
- Islam, R.; Singh, V.; Ammeter, D.; Schweizer, F.; Kuss, S. Electrochemical Characterization of the Antibiotic Hybrid Ciprofloxacin-Tobramycin. Electrochem. Commun. 2020, 119, 106825.
- Seiler, P.; Enderlin-Paput, M.; Pfaff, P.; Weiss, M.; Ritz, D.; Clozel, M.; Locher, H.H. Cadazolid Does Not Promote Intestinal Colonization of Vancomycin-Resistant Enterococci in Mice. Antimicrob. Agents Chemother. 2015, 60, 628–631.
- Muhammad, A.; Simcha, W.; Rawish, F.; Sabih, R.; Albert, E.; Ali, N. Cadazolid vs Vancomycin for the Treatment of Clostridioides Difficile Infection: Systematic Review with Meta-Analysis. Curr. Clin. Pharmacol. 2020, 15, 4–10.
- Wang, Y.; Damu, G.L.V.; Lv, J.-S.; Geng, R.-X.; Yang, D.-C.; Zhou, C.-H. Design, Synthesis and Evaluation of Clinafloxacin Triazole Hybrids as a New Type of Antibacterial and Antifungal Agents. Bioorg. Med. Chem. Lett. 2012, 22, 5363–5366.
- Eissa, S.I.; Farrag, A.M.; Abbas, S.Y.; El Shehry, M.F.; Ragab, A.; Fayed, E.A.; Ammar, Y.A. Novel Structural Hybrids of Quinoline and Thiazole Moieties: Synthesis and Evaluation of Antibacterial and Antifungal Activities with Molecular Modeling Studies. Bioorganic Chem. 2021, 110, 104803.
- Kowalczuk, D.; Gładysz, A.; Pitucha, M.; Kamiński, D.M.; Barańska, A.; Drop, B. Spectroscopic Study of the Molecular Structure of the New Hybrid with a Potential Two-Way Antibacterial Effect. Molecules 2021, 26, 1442.
- Zhang, F.Y.; Du, G.J.; Zhang, L.; Zhang, C.L.; Lu, W.L.; Liang, W. Naringenin Enhances the Anti-Tumor Effect of Doxorubicin through Selectively Inhibiting the Activity of Multidrug Resistance-Associated Proteins but Not P-Glycoprotein. Pharm. Res. 2009, 26, 914–925.
- Hryhoriv, H.; Mariutsa, I.; Kovalenko, S.M.; Georgiyants, V.; Perekhoda, L.; Filimonova, N.; Geyderikh, O.; Sidorenko, L. The Search for New Antibacterial Agents among 1,2,3-Triazole Functionalized Ciprofloxacin and Norfloxacin Hybrids: Synthesis, Docking Studies, and Biological Activity Evaluation. Sci. Pharm. 2022, 90, 2.
- Albayrak, F.; Çiçek, M.; Alkaya, D.; Kulu, I. Design, Synthesis and Biological Evaluation of 8-Aminoquinoline-1,2,3-Triazole Hybrid Derivatives as Potential Antimicrobial Agents. Med. Chem. Res. 2022, 31, 652–665.
- Awolade, P.; Cele, N.; Ebenezer, O.; Kerru, N.; Gummidi, L.; Gu, L.; Palma, G.; Kaur, M.; Singh, P. Synthesis of 1H-1,2,3-Triazole-Linked Quinoline-Isatin Molecular Hybrids as Anti-Breast Cancer and Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Agents. Anticancer Agents Med. Chem. 2021, 21, 1228–1239.
- Millanao, A.R.; Mora, A.Y.; Villagra, N.A.; Bucarey, S.A.; Hidalgo, A.A. Biological Effects of Quinolones: A Family of Broad-Spectrum Antimicrobial Agents. Molecules 2021, 26, 7153.
- Hu, Y.-Q.; Gao, C.; Zhang, S.; Xu, L.; Xu, Z.; Feng, L.-S.; Wu, X.; Zhao, F. Quinoline Hybrids and Their Antiplasmodial and Antimalarial Activities. Eur. J. Med. Chem. 2017, 139, 22–47.
- Fan, Y.-L.; Cheng, X.-W.; Wu, J.-B.; Liu, M.; Zhang, F.-Z.; Xu, Z.; Feng, L.-S. Antiplasmodial and Antimalarial Activities of Quinolone Derivatives: An Overview. Eur. J. Med. Chem. 2018, 146, 1–14.
- Abdel-Aziz, M.; Park, S.-E.; Abuo-Rahma, G.E.-D.A.A.; Sayed, M.A.; Kwon, Y. Novel N-4-Piperazinyl-Ciprofloxacin-Chalcone Hybrids: Synthesis, Physicochemical Properties, Anticancer and Topoisomerase I and II Inhibitory Activity. Eur. J. Med. Chem. 2013, 69, 427–438.
- Fallica, A.N.; Barbaraci, C.; Amata, E.; Pasquinucci, L.; Turnaturi, R.; Dichiara, M.; Intagliata, S.; Gariboldi, M.B.; Marras, E.; Orlandi, V.T.; et al. Nitric Oxide Photo-Donor Hybrids of Ciprofloxacin and Norfloxacin: A Shift in Activity from Antimicrobial to Anticancer Agents. J. Med. Chem. 2021, 64, 11597–11613.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
3 times
(View History)
Update Date:
22 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No