 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shiva Moein | -- | 1657 | 2022-09-07 03:50:20 | | | |
| 2 | Beatrix Zheng | + 5 word(s) | 1662 | 2022-09-19 03:21:50 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 1662 | 2022-09-19 08:24:58 | | | | |
| 4 | Beatrix Zheng | Meta information modification | 1662 | 2022-09-20 08:44:42 | | |
Video Upload Options
Spalt-Like Transcription Factor 4 (SALL4) is a critical factor for self-renewal ability and pluripotency of stem cells. Also, Various reports show tight relation of SALL4 to cancer occurrence and metastasis. SALL4 exerts its effects not only by inducing gene expression but also by repressing a large cluster of genes through interaction with various epigenetic modifiers. Due to the high expression of this protein in cancer cells and its silence in almost all adult tissues, it is an ideal target for cancer therapy.
1. Introduction
2. SALL4 as a Potential Therapeutic Target
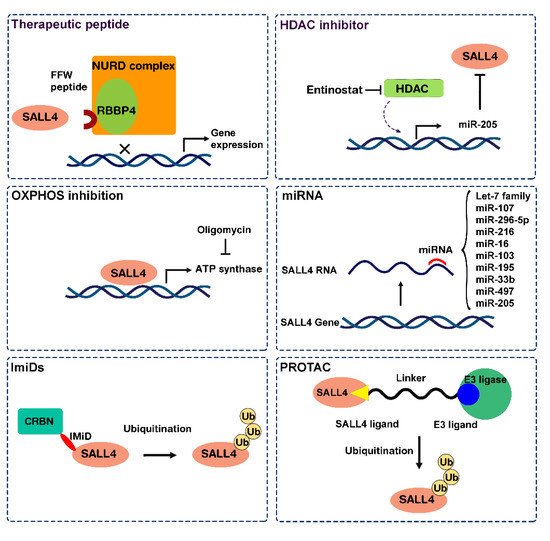
2.1. Approach 1: Target SALL4 Function, the SALL4 Inhibitors
2.2. Approach 2: Targeting SALL4 Downstream Pathways; Repurposing Oxidative Phosphorylation Drugs to Inhibit SALL4 Positive Cells
2.3. Approach 3: Modulating SALL4 Abundancy
2.3.1. Nucleic Acid-Based Therapy
2.3.2. SALL4 Degraders
3. Conclusion
Despite the evident role of SALL4 in tumorigenesis and therapy resistance, no therapeutic targeting SALL4 are currently on the market. The landmark discovery of a new class of molecular glues non-IMiD targeting SALL4 and other oncogenes has the potential to become a real disease-modifying treatment and open a novel therapeutic perspective for several intractable cancer conditions.
References
- Elling, U.; Klasen, C.; Eisenberger, T.; Anlag, K.; Treier, M.; Murine inner cell mass-derived lineages depend on Sall4 function. Proceedings of the National Academy of Sciences of the United States of America 2006, 103, 16319-16324, doi:10.1073/pnas.0607884103.
- Kohlhase, J.; Chitayat, D.; Kotzot, D.; Ceylaner, S.; Froster, U.G.; Fuchs, S.; Montgomery, T.; Rösler, B. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Human mutation 2005, 26, 176-183, doi:10.1002/humu.20215.
- Yang, J.J.B.R. SALL4 as a transcriptional and epigenetic regulator in normal and leukemic hematopoiesis. 2018, 6, 1-9.4. Miettinen, M.; Wang, Z.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Biernat, W.; Lasota, J.; Lee, Y.-S. SALL4 expression in germ cell and non-germ cell tumors: a systematic immunohistochemical study of 3215 cases. Am J Surg Pathol 2014, 38, 410-420, doi:10.1097/PAS.0000000000000116.
- Kühnlein, R.P.; Frommer, G.; Friedrich, M.; Gonzalez-Gaitan, M.; Weber, A.; Wagner-Bernholz, J.F.; Gehring, W.J.; Jäckle, H.; Schuh, R. spalt encodes an evolutionarily conserved zinc finger protein of novel structure which provides homeotic gene function in the head and tail region of the Drosophila embryo. EMBO J 1994, 13, 168-179, doi:10.1002/j.1460-2075.1994.tb06246.x.
- Fujii, Y.; Yoshihashi, K.; Suzuki, H.; Tsutsumi, S.; Mutoh, H.; Maeda, S.; Yamagata, Y.; Seto, Y.; Aburatani, H.; Hatakeyama, M. CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 20584-20589, doi:10.1073/pnas.1208651109.
- Nicolè, L.; Sanavia, T.; Veronese, N.; Cappellesso, R.; Luchini, C.; Dabrilli, P.; Fassina, A. Oncofetal gene SALL4 and prognosis in cancer: A systematic review with meta-analysis. Oncotarget 2017, 8, 22968-22979, doi:10.18632/oncotarget.14952
- Liu, C.; Yao, F.; Mao, X.; Li, W.; Chen, H. Effect of SALL4 on the Proliferation, Invasion and Apoptosis of Breast Cancer Cells. Technology in cancer research & treatment 2020, 19, 1533033820980074, doi:10.1177/1533033820980074.Di, C.; Sun, J.; Zhang, H.; Zhou, P.; Kong, J. High expression of SALL4 is associated with poor prognosis in squamous cell carcinoma of the uterine cervix. Int J Clin Exp Pathol 2018, 11, 1391-1398
- Han, S.X.; Wang, J.L.; Guo, X.J.; He, C.C.; Ying, X.; Ma, J.L.; Zhang, Y.Y.; Zhao, Q.; Zhu, Q. Serum SALL4 is a novel prognosis biomarker with tumor recurrence and poor survival of patients in hepatocellular carcinoma. Journal of immunology research 2014, 2014, 262385, doi:10.1155/2014/262385.
- Sun, B.; Xu, L.; Bi, W.; Ou, W.-B. SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance. Int J Mol Sci 2022, 23, 2053, doi:10.3390/ijms23042053
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Research 2018, 28, 265-280, doi:10.1038/cr.2017.155
- Liu, B.H.; Jobichen, C.; Chia, C.S.B.; Chan, T.H.M.; Tang, J.P.; Chung, T.X.Y.; Li, J.; Poulsen, A.; Hung, A.W.; Koh-Stenta, X.; et al. Targeting cancer addiction for SALL4 by shifting its transcriptome with a pharmacologic peptide. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, E7119-e7128, doi:10.1073/pnas.1801253115
- Yong, K.J.; Li, A.; Ou, W.B.; Hong, C.K.; Zhao, W.; Wang, F.; Tatetsu, H.; Yan, B.; Qi, L.; Fletcher, J.A.; et al. Targeting SALL4 by entinostat in lung cancer. Oncotarget 2016, 7, 75425-75440, doi:10.18632/oncotarget.12251
- Tan, J.L.; Li, F.; Yeo, J.Z.; Yong, K.J.; Bassal, M.A.; Ng, G.H.; Lee, M.Y.; Leong, C.Y.; Tan, H.K.; Wu, C.S.; et al. New High-Throughput Screening Identifies Compounds That Reduce Viability Specifically in Liver Cancer Cells That Express High Levels of SALL4 by Inhibiting Oxidative Phosphorylation. Gastroenterology 2019, 157, 1615-1629.e1617, doi:10.1053/j.gastro.2019.08.022
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Research 2018, 28, 265-280, doi:10.1038/cr.2017.155.91. Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Frontiers in genetics 2017, 8, 31, doi:10.3389/fgene.2017.00031
- Jin, B.; Wang, W.; Meng, X.-x.; Du, G.; Li, J.; Zhang, S.-z.; Zhou, B.-h.; Fu, Z.-h. Let-7 inhibits self-renewal of hepatocellular cancer stem-like cells through regulating the epithelial-mesenchymal transition and the Wnt signaling pathway. BMC Cancer 2016, 16, 863, doi:10.1186/s12885-016-2904-y.
- Shen, C.; Li, J.; Che, G.J.T.C.R. Prognostic value of let-7 in lung cancer: systematic review and meta-analysis. 2020 2020, 9, 6354-6361.
- Xia, Z.; Qiu, D.; Deng, J.; Jiao, X.; Yang, R.; Sun, Z.; Wan, X.; Li, J. Methylation-induced downregulation and tumor-suppressive role of microRNA-98 in glioma through targeting Sal-like protein 4. International journal of molecular medicine 2018, 41, 2651-2659, doi:10.3892/ijmm.2018.3464
- Shi, D.M.; Shi, X.L.; Xing, K.L.; Zhou, H.X.; Lu, L.L.; Wu, W.Z. miR-296-5p suppresses stem cell potency of hepatocellular carcinoma cells via regulating Brg1/Sall4 axis. Cellular signalling 2020, 72, 109650, doi:10.1016/j.cellsig.2020.109650.
- Chen, L.P.; Zhang, N.N.; Ren, X.Q.; He, J.; Li, Y. miR-103/miR-195/miR-15b Regulate SALL4 and Inhibit Proliferation and Migration in Glioma. Molecules (Basel, Switzerland) 2018, 23, doi:10.3390/molecules23112938.
- Liu, J.; Sauer, M.A.; Hussein, S.G.; Yang, J.; Tenen, D.G.; Chai, L. SALL4 and microRNA: The Role of Let-7. Genes 2021, 12, doi:10.3390/genes12091301
- Hesari, A.; Anoshiravani, A.A.; Talebi, S.; Noruzi, S.; Mohammadi, R.; Salarinia, R.; Zare, R.; Ghasemi, F. Knockdown of sal-like 4 expression by small interfering RNA induces apoptosis in breast cancer cells. Journal of cellular biochemistry 2019, 120, 9392-9399, doi:10.1002/jcb.28214.
- Ashrafizadeh, M.; Hushmandi, K.; Rahmani Moghadam, E.; Zarrin, V.; Hosseinzadeh Kashani, S.; Bokaie, S.; Najafi, M.; Tavakol, S.; Mohammadinejad, R.; Nabavi, N.; et al. Progress in Delivery of siRNA-Based Therapeutics Employing Nano-Vehicles for Treatment of Prostate Cancer. 2020, 7, 91.
- Yanagihara, N.; Kobayashi, D.; Kuribayashi, K.; Tanaka, M.; Hasegawa, T.; Watanabe, N. Significance of SALL4 as a drug‑resistant factor in lung cancer. International journal of oncology 2015, 46, 1527-1534, doi:10.3892/ijo.2015.2866.
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Słabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thomä, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572, doi:10.1126/science.aat0572.
- Donovan, K.A.; An, J.; Nowak, R.P.; Yuan, J.C.; Fink, E.C.; Berry, B.C.; Ebert, B.L.; Fischer, E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 2018, 7, doi:10.7554/eLife.38430.
- Maneiro, M.; De Vita, E.; Conole, D.; Kounde, C.; Zhang, Q.; Tate, E.J.P.i.m.c. PROTACs, molecular glues and bifunctionals from bench to bedside: Unlocking the clinical potential of catalytic drugs. 2021, 60, 67-190.


