 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cinta Pegueroles | + 2799 word(s) | 2799 | 2020-10-15 08:18:36 | | | |
| 2 | Dean Liu | -1034 word(s) | 1765 | 2020-10-22 02:50:49 | | | | |
| 3 | Dean Liu | Meta information modification | 1765 | 2020-10-27 08:05:31 | | |
Video Upload Options
Colorectal cancer is the third most common cause of cancer mortality worldwide, and one of the current hot topics in this field is the identification of markers for identifying patients with high probability of relapse, which may benefit from adjuvant chemotherapy in stage II. In this sense long noncoding RNAs are a promising source of novel cancer biomarkers and therapeutic targets, although to robustly assess their gene expression levels is challenging due to their low expression. We present a protocol (hereafter referred as CoLong design) which couples target enrichment and RNA-seq that allows transcriptomics studies of lncRNAs in formalin-fixed paraffin embedded (FFPE) tissue samples. We show that our new approach alllows to efficiently detect differences in gene expression and somatic mutations in lncRNAs, and we propose several lncRNAs as potential candidates for colorectal cancer. This new approach could represent a promising avenue that would reduce costs and enable more efficient translational research.
1. Introduction
Long non-coding RNAs (lncRNAs) are a large and heterogeneous class of transcripts that are longer than 200 nucleotides, and have limited coding potential[1][2]. Most lncRNAs are transcribed by RNA polymerase II, capped, spliced, and polyadenylated[3]. However, compared to protein-coding mRNAs, lncRNAs are shorter, have fewer exons, are expressed at lower levels and in a more tissue- and cell-specific manner, and are alternatively spliced more frequently[4]. From an evolutionary standpoint, lncRNAs are poorly conserved, which limits their study in model organisms[5]. Accumulating evidence supports the involvement of lncRNAs in all aspects of gene regulation and, accordingly, numerous lncRNAs have been linked to diverse human diseases, including cancer[6][7]. For instance, transcriptomic analyses have shown that changes in the expression of lncRNAs are among the most commonly observed alterations in cancer[8][9]. Although the role of lncRNAs is still poorly understood, their involvement in cancer paves the way to novel therapeutic, diagnostic, or prognostic approaches centered on these molecules. We used a new tool (based on target enrichment and RNA-seq), which allows transcriptomics studies of low expressed molecules such as lncRNAs in formalin-fixed paraffin embedded (FFPE) tissue samples.
2. Identification of Differentially Expressed lncRNAs in CRC
We next set out to find lncRNAs that are differentially expressed (DE) in CRC tumors by applying our validated methodology (CoLong design) to FFPE samples. To detect DE lncRNAs, we used samples from 35 individuals with stage II CRC, which passed quality filters and had paired samples from tumoral tissue and the surrounding healthy tissue—referred to as normal hereafter. Different clustering approaches showed that expression profiles of lncRNAs separated samples in function of the disease condition, with an additional effect of sex (Figure 1).
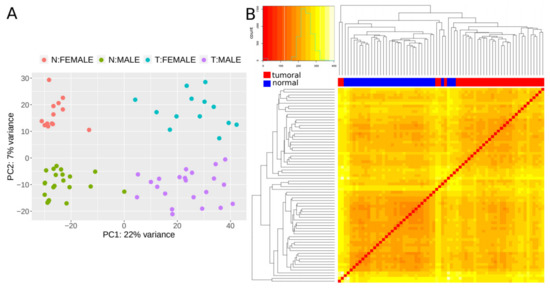
Figure 1. Expression patterns of lncRNAs. (A) Principal component analysis (PCA) plot based on transformed count data after variance stabilizing transformation. T and N indicate tumoral and normal samples, respectively. (B) Heatmap showing the overall similarity between samples based on a hierarchical clustering of the Euclidean distance between samples using the variance stabilizing transformation (vst-transformed) data. Red and blue bars at the top of the columns designate tumoral and normal samples, respectively.
Then, using a multi-factor design that considers ‘individual’ and ‘condition’ as independent variables, we identified 379 lncRNAs out of the 7,812 lncRNAs included in the final dataset that were DE between normal and tumoral samples, with 48.8% and 51.2% of them being up- and down-regulated respectively (Figure 2A). Importantly, most of the samples showed the same directionality, as indicated by lines connecting normal and tumoral samples for a given individual (Figure 2B).
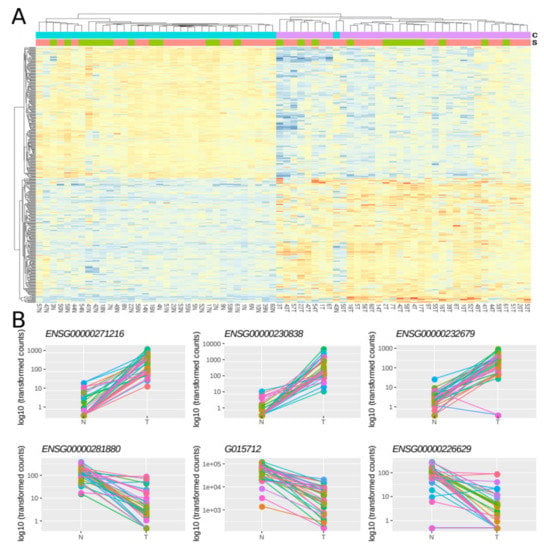
Figure 2. Differentially expressed lncRNAs in FFPE CRC stage II samples. (A) Heatmap showing gene expression from the 379 DE lncRNAs. Color gradient ranges from low expressed (blue) to highly expressed (red). (B) Dot plots showing normalized count data in a logarithmic scale of normal and tumoral samples for the three top up- and down-DE lncRNAs. Horizontal bars on top show condition (blue for normal, pink for tumoral) and sex (green for female and red for male). Each individual is represented with a different color, lines group paired samples from the same individual and T and N indicate tumoral and normal samples, respectively.
To compare the results of the enrichment-based FFPE procedure with those of the non-enrichment FF procedure, we downloaded RNAseq data from 40 matched pairs of samples from a CRC study available in The Cancer Genome Atlas (TCGA)[10]. Using the same procedure on this dataset as that described above, we identified 247 DE lncRNAs between normal and tumoral samples. Despite the different cohorts, nature of the samples, and protocols, we found that the comparisons had a significant overlap of 130 DE lncRNAs, which were similarly up- or down-regulated in the two studies. Of note, the enrichment-based FFPE dataset found a higher number of DE lncRNAs as compared to the FF study (379 vs. 247) and missed a lower fraction of DE lncRNA from the non-enriched dataset (117 out of 247) than the reverse comparison (249 out of 379). This observation suggests that differences are partly due to a greater ability of the enrichment-based approach to detect DE lncRNAs.
We examined whether DE lncRNAs identified exclusively in one of the two studies shared characteristics that may explain these discrepancies. In this regard, lncRNAs exclusively detected as DE in FFPE had lower expression levels, thereby suggesting that the absence of lncRNA enrichment in the TCGA study may have limited the analyses of lncRNAs with low expression. This notion is further supported by the observation that lncRNAs that were DE exclusively in the FFPE dataset showed lower expression values in data mined from several public repositories (Human Body Map, Array Express, ENCODE, etc) included in LnCompare[11].
We next assessed further characteristics of the lncRNAs detected as DE by each of the approaches and the overlap of the two. All three lists of DE lncRNA were enriched in functional lncRNAs (10% for FFPE, 13.7% for TCGA, and 17.7% for the overlap) compared to 3.2% functional lncRNAs in the CoLong design (p-value ≤ 1 × 10−9 after the hypergeometric test in all cases). The three lists were also enriched in genes known to be involved in CRC (9% for FFPE, 13% for TCGA, and 17.7% for the overlap; p-value < 2 × 10−10 hypergeometric test in all cases. Similarly, we found that few DE lncRNAs had coding potential (5% of FFPE, 6.5% of TCGA and 5.4% of overlap), being candidates to encode for small peptides. We found that 43%, 39%, and 43% of DE lncRNAs in the FFPE, TCGA, and overlap lists, respectively, have evolutionarily conserved splice sites, which was similar for the bulk of all targeted genes (40.5%). The FEZF1-AS1 gene, which was DE in the three lists, contains an ultraconserved element (uc232), which has been proposed to be involved in cancer[11]. However, its location overlaps with a protein-coding gene involved in cancer (CADPS). Finally, when comparing other characteristics of the lists of DE lncRNAs with GENCODE v24 lncRNAs[3], we found that the three lists were significantly enriched in functionally validated and disease associated lncRNAs, lncRNAs containing retroviral elements repeats (ERVL-MaLR,ERV1), lncRNAs genes with longer total and exonic lengths, lncRNAs with protein-coding genes upstream in the same strand, and with lower distances to the closest protein coding gene (adjusted p-value < 0.05). We searched for experimentally validated interactions of DE lncRNAs in the NPInter v4.0 database[12], and found that 82, 62, and 29 lncRNAs in the FFPE, TCGA, and overlap lists, respectively, interacted with RNAs or proteins. We performed a functional enrichment analysis of the set of interacting proteins and found significant enrichment in several biological processes (adjusted p-value < 0.05). Interestingly, some of the genes with interactions had no functional annotation in the databases, for instance, ENSG00000231881 and ENSG00000233858). For ENSG00000231881, we found a recent study suggesting that it regulates the VEGF signaling pathway by binding to miR-133b, thereby promoting metastasis in CRC cells[12][13]. This notion is consistent with our results showing that ENSG00000231881 is up-regulated in CRC tumors. Regarding ENSG00000233858, we found that it is downregulated in tumoral samples. This observation contrasts with a recent study that found that this gene was highly expressed in metastatic breast cancer, and associated it with poor prognosis[14]. That study concluded that ENSG00000233858 and UCA1 (ENSG00000214049) could cooperatively upregulate the expression of the Slug gene, which has been associated with poor prognosis and survival in CRC[15][16][17]. Interestingly, UCA1 is significantly up-regulated in tumors in our analysis. Thus, the interplay of UCA1 and ENSG00000233858 is complex and may also be involved in CRC.
All together, our results underscore the power of a probe-based enrichment approach to assess DE lncRNAs in FFPE samples, and provide a candidate list of lncRNAs likely to be involved in CRC. In particularly, the 130 lncRNAs detected as DE in both the TCGA and FFPE datasets are strong candidates to be involved in CRC, as this set is significantly enriched in disease-related genes and in genes previously associated with this malignancy.
3. Identification of CRC-Related Somatic Mutations in FFPE Samples
Genomic alterations are common in CRC and act as drivers of tumorigenesis[18]. Chromosomal instability, and variations in microsatellites and Cytosine-phosphate-Guanine (CpG) islands have been widely studied in the context of CRC, but little is known about somatic variants in lncRNAs. To date, only seven single nucleotide polymorphisms (SNPs) in lncRNAs have been associated with CRC[19]. Thus, we studied the presence of putative somatic mutations in lncRNAs using our RNAseq data from FFPE samples. We detected a higher number of putative somatic mutations in tumoral than in normal tissues (4,995 and 1,422, respectively). This finding is consistent with a previous study that concluded that CRC cells show a substantial increase in somatic mutation rate compared to normal colorectal cells[20]. Most somatic variants were present in a single individual (89.7% and 79.1% in tumoral and normal samples respectively), while variants shared by both tissues from an individual were commonly present in several individuals. Of note, we detected 184 somatic mutations shared by three or more individuals, and 11 of these were common and present in eight or more individuals. These eleven frequent CRC somatic variants affected eight lncRNAs (ENSG00000226476, ENSG00000281406 (BLACAT1), ENSG00000248323 (LUCAT1), ENSG00000271762, ENSG00000282961 (PNRCR1), ENSG00000253929 (CASC21), ENSG00000245532 (NEAT1), ENSG00000265975), of which the following five had a known role in CRC: BLACAT1 [21][22]; LUCAT1[23]; PNRCR1[24]; CASC21[25]; and NEAT1[26]. In addition, four of these commonly mutated lncRNAs were DE in both the FFPE and TCGA datasets (BLACAT1, LUCAT1, ENSG00000253929, and ENSG00000226476). Our set of CRC somatic mutations includes one of the seven already described (rs7013433 in CCAT1), for which we found the same alternative allele reported in dbSNP[27]). Our dataset includes eight individuals who relapsed and we found that 21 of the CRC somatic variants showed higher frequency in relapsed as compared with non-relapsed. Four lncRNAs (ENSG0000021403, ENSG00000261650, ENSG00000281406, G001643) accumulated multiple such relapse-associated variants. These results suggest a prognostic value for those somatic variants and a possible role of these lncRNAs in cancer progression.
References
- Brown, C.J.; Hendrich, B.; Rupert, J.L.; Lafrenière, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542.
- Clemson, C.M.; A McNeil, J.; Willard, H.F.; Lawrence, J.B. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Boil. 1996, 132, 259–275.
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martín, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789.
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.J.; Tazon-Vega, B.; Regev, A.; Rinn, J. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927.
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641.
- Deniz, E.; Erman, B. Long noncoding RNA (lincRNA), a new paradigm in gene expression control. Funct. Integr. Genom. 2016, 17, 135–143.
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509.
- Du, Z.; Fei, T.; Verhaak, R.G.; Su, Z.; Zhang, Y.; Brown, M.; Chen, Y.; Liu, X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Boil. 2013, 20, 908–913.
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208.
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Carlevaro-Fita, J.; Liu, L.; Zhou, Y.; Zhang, S.; Chouvardas, P.; Johnson, R.; Li, J. LnCompare: gene set feature analysis for human long non-coding RNAs. Nucleic Acids Res. 2019, 47, W523–W529.
- Fiorenzano, A.; Pascale, E.; Gagliardi, M.; Terreri, S.; Papa, M.; Andolfi, G.; Galasso, M.; Tagliazucchi, G.M.; Taccioli, C.; Patriarca, E.J.; et al. An Ultraconserved Element Containing lncRNA Preserves Transcriptional Dynamics and Maintains ESC Self-Renewal. Stem Cell Rep. 2018, 10, 1102–1114.
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165.
- Zhou, J.; Zou, Y.; Hu, G.; Lin, C.; Guo, Y.; Gao, K.; Wu, M. Facilitating colorectal cancer cell metastasis by targeted binding of long non-coding RNA ENSG00000231881 with miR-133b via VEGFC signaling pathway. Biochem. Biophys. Res. Commun. 2019, 509, 1–7.
- Li, G.-Y.; Wang, W.; Sun, J.-Y.; Xin, B.; Zhang, X.; Wang, T.; Zhang, Q.-F.; Yao, L.-B.; Han, H.; Fan, D.-M.; et al. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018, 8, 2846–2861.
- Shioiri, M.; Shida, T.; Koda, K.; Seike, K.; Nishimura, M.; Takano, S.; Miyazaki, M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br. J. Cancer 2006, 94, 1816–1822.
- Du, L.; Li, J.; Lei, L.; He, H.; Chen, E.; Dong, J.; Yang, J. High Vimentin Expression Predicts a Poor Prognosis and Progression in Colorectal Cancer: A Study with Meta-Analysis and TCGA Database. BioMed Res. Int. 2018, 2018, 1–14.
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Goel, A.; Kusunoki, M. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinog. 2013, 34, 2548–2557.
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest Cancer Res 2012, 5, 19–27.
- Minotti, L.; Agnoletto, C.; Baldassari, F.; Corrà, F.; Volinia, S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput 2018, 7, 34.
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462.
- Lu, H.; Liu, H.; Yang, X.; Ye, T.; Lv, P.; Wu, X.; Ye, Z. LncRNA BLACAT1 May Serve as a Prognostic Predictor in Cancer: Evidence from a Meta-Analysis. BioMed Res. Int. 2019, 2019, 1275491.
- Gao, X.; Wen, J.; Gao, P.; Zhang, G.; Zhang, G. Overexpression of the long non-coding RNA, linc-UBC1, is associated with poor prognosis and facilitates cell proliferation, migration, and invasion in colorectal cancer. OncoTargets Ther. 2017, 10, 1017–1026.
- Zhou, Q.; Hou, Z.; Zuo, S.; Zhou, X.; Feng, Y.; Sun, Y.; Yuan, X. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40- MDM 2-p53 pathway through binding with UBA 52. Cancer Sci. 2019, 110, 1194–1207.
- Yang, L.; Qiu, M.; Xu, Y.; Wang, J.; Zheng, Y.; Li, M.; Xu, L.; Yin, R. Upregulation of long non-coding RNA PRNCR1 in colorectal cancer promotes cell proliferation and cell cycle progression. Oncol. Rep. 2015, 35, 318–324.
- Zheng, Y.; Nie, P.; Xu, S. Long noncoding RNA CASC21 exerts an oncogenic role in colorectal cancer through regulating miR-7-5p/YAP1 axis. Biomed. Pharmacother. 2020, 121, 109628.
- Idogawa, M.; Ohashi, T.; Sasaki, Y.; Nakase, H.; Tokino, T. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int. J. Cancer 2017, 140, 2785–2791.

