Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gerald I Nwosu | -- | 1387 | 2022-09-13 17:10:36 | | | |
| 2 | Jessie Wu | + 6 word(s) | 1393 | 2022-09-14 03:36:26 | | | | |
| 3 | Jessie Wu | Meta information modification | 1393 | 2022-09-14 03:45:09 | | | | |
| 4 | Jessie Wu | Meta information modification | 1393 | 2022-09-15 08:05:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nwosu, G.; Reddy, S.B.; Riordan, H.R.M.; Kang, J. Pathomechanisms for GABRG2 and GABRB3 Mutations in DEEs. Encyclopedia. Available online: https://encyclopedia.pub/entry/27136 (accessed on 03 August 2026).
Nwosu G, Reddy SB, Riordan HRM, Kang J. Pathomechanisms for GABRG2 and GABRB3 Mutations in DEEs. Encyclopedia. Available at: https://encyclopedia.pub/entry/27136. Accessed August 03, 2026.
Nwosu, Gerald, Shilpa B. Reddy, Heather Rose Mead Riordan, Jing-Qiong Kang. "Pathomechanisms for GABRG2 and GABRB3 Mutations in DEEs" Encyclopedia, https://encyclopedia.pub/entry/27136 (accessed August 03, 2026).
Nwosu, G., Reddy, S.B., Riordan, H.R.M., & Kang, J. (2022, September 13). Pathomechanisms for GABRG2 and GABRB3 Mutations in DEEs. In Encyclopedia. https://encyclopedia.pub/entry/27136
Nwosu, Gerald, et al. "Pathomechanisms for GABRG2 and GABRB3 Mutations in DEEs." Encyclopedia. Web. 13 September, 2022.
Copy Citation
GABAA receptor genes (GABR) are a group of genes associated with DEE, although previous studies have been focused on relatively mild epileptic syndromes, such as childhood absence epilepsy (CAE), febrile seizures (FS), and generalized tonic-clonic seizures with febrile seizure plus (GEFS+). Mutations in GABAA receptor subunit genes (GABRs) are a major etiology for developmental and epileptic encephalopathies (DEEs).
GABRB3
GABRB2
DEEs
1. Pathomechanisms for GABRG2 Mutations in Developmental and Epileptic Encephalopathies
The most common phenotypes associated with GABRG2 mutations are febrile seizures, generalized tonic-clonic seizures with febrile plus (GEFS+), and DS, with febrile seizures being the core phenotype (Figure 1A). Extensive work from previous studies has shown that GABRG2 mutations are associated with a wide spectrum of seizure disorders (Figure 1B). There has been much investigation into the impact of GABRG2 mutations in vitro and in vivo using knock-in mouse models, which would allow researchers to infer the pathophysiology of this mutation in the patient.
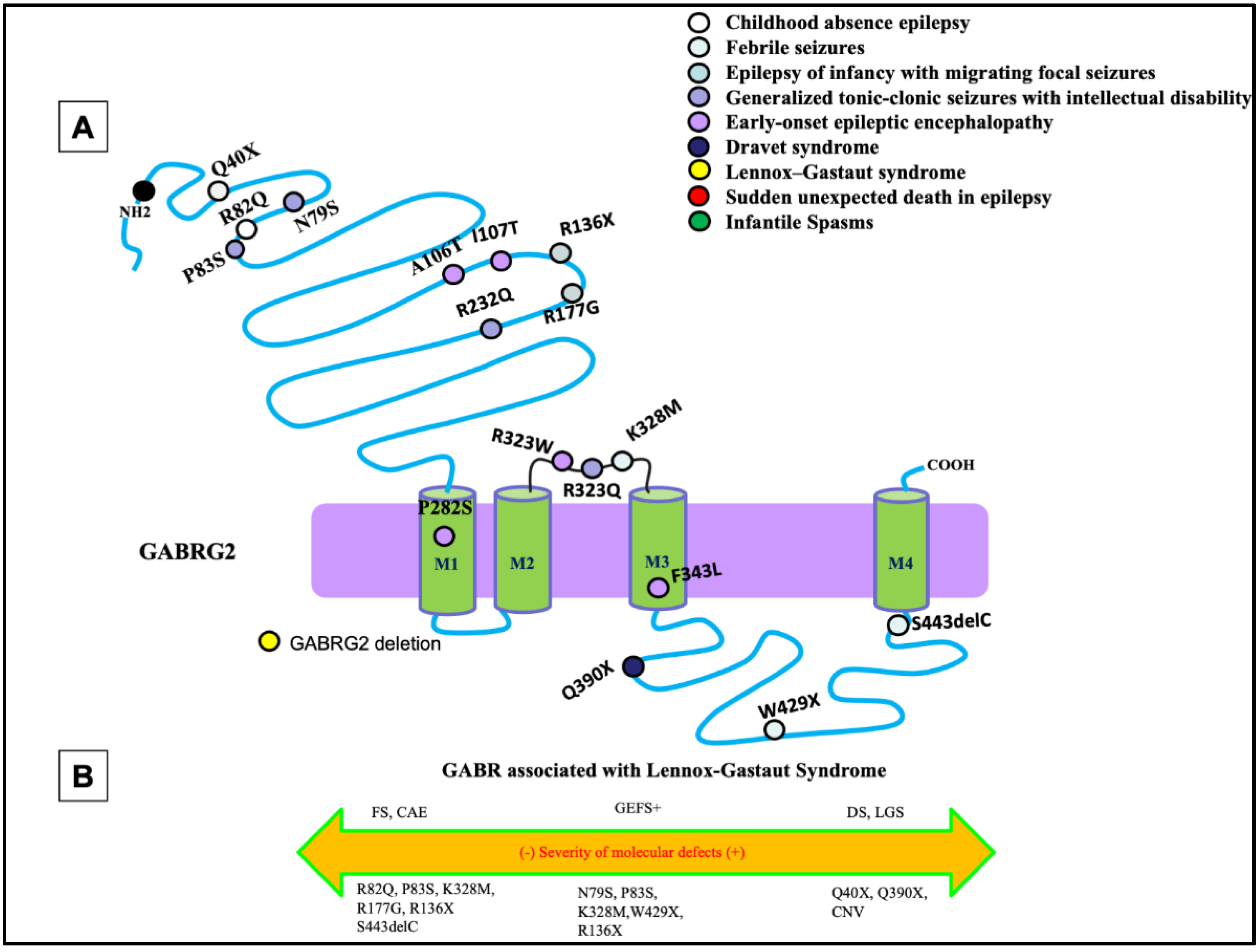
Figure 1. Epilepsy phenotypes associated with GABRG2 mutations. (A) Two-dimensional protein topology of the γ2 subunit showing locations of mutations in human GABRG2 associated with various epilepsy syndromes and neurodevelopmental disorders. Each mutation is color coded to indicate associated diagnosis, and each dot represents the relative location on the γ2 subunit protein peptide. (B) Diagram dictating the severity of each mutation on a continuum of epilepsy phenotypes. The left side of the diagram infers a less severe epileptic phenotype, the middle dictates a moderate, and the right side of the diagram infers a more severe epileptic phenotype.
Mutations of GABRG2 are frequently associated with developmental and epileptic disorders through both missense and nonsense mutations. Nonsense mutations in GABRG2 have been identified and are associated with FS, GEFS+, or DEEs. A GABRG2 (R43Q) mutation had reduced cell surface expression of the subunit and reduced cortical inhibition in knock-in mice [1][2]. However, in some cases those mutant receptors, which traffic to the cell surface, have normal function [3]. It was also identified that a subset of mutations, especially the nonsense mutations GABRG2 (Q351X), cannot traffic to the cell surface and synapse due to severe misfolding [4][5]. The mutant protein was retained in the endoplasmic reticulum and reduced trafficking of wildtype partnering subunits, implicating its dominant-negative suppression causing GEFS+. The mutant GABRG2 (Q351X) has slow degradation and can impose a dominant-negative effect on the wildtype GABRG2 allele and the partnering subunits [4]. These findings have been validated in vivo with Gabrg2+/Q390X mice, showing that the mutation GABRG2 (Q390X) is proven to cause chronic subunit accumulation and neurodegeneration [5]. In older mice, the mutated subunit was shown to form protein aggregates due to chronic accumulation of the mutant protein in the endoplasmic reticulum [5]. Mice expressing the GABRG2 (Q390X) mutation showed a severe epileptic phenotype, which included spontaneous generalized tonic-clonic seizures in a seizure-resistant C57BL/6J mouse background. The phenotype is much more severe than the Gabrg2+/− heterozygous knockout mice without generation of the mutant protein [6].
As mentioned above, the γ2 subunit plays an essential role in GABAergic neurotransmission and the overall homeostasis of the central nervous system, while impairment in the subunit results in seizures and neurodevelopmental abnormalities. Some mutants of this protein display a dominant-negative effect that suppresses expression of both wildtype subunit and its binding partners in the hetero-pentamer. Of note, the proband displays an uncharacteristic phenotype for a mutation of this subunit, suggesting further investigation into the interplay of associated proteins with the receptor and how their function is affected by the mutation. The GABAA receptor exerts channel function as a pentamer, and the γ2 subunits oligomerize and colocalize with partnering subunits. There is the possibility that the deficiency due to mutant γ2 protein can result in suppression of proper function of the β3 subunit whose perturbation is often attributed to the LGS phenotype.
2. Pathomechanisms for GABRB3 Mutations in Developmental and Epileptic Encephalopathies
Like the phenotypic heterogeneity associated with mutations in GABRG2, mutations in GABRB3 have also been associated with a spectrum of disease phenotypes, including ASD, IS, and DS [7][8] (Figure 2A,B). Among all GABRs, only mutations in GABRB3 have been previously reported to be associated with LGS [9]. In addition to LGS, the β3 subunit encoded by GABRB3 has also been frequently associated with CAE [10][11]. In addition to the defect in the mutant β3 subunit, a mutant β3 subunit can also compromise the assembly and trafficking of partnering subunits α1 or γ2. This suggests there may exist overlapping phenotypes at the clinical level among mutations in the partnering subunits of the receptor through a dominant-negative effect of one, suppressing the maturation and trafficking of the others, as previously reported [6].
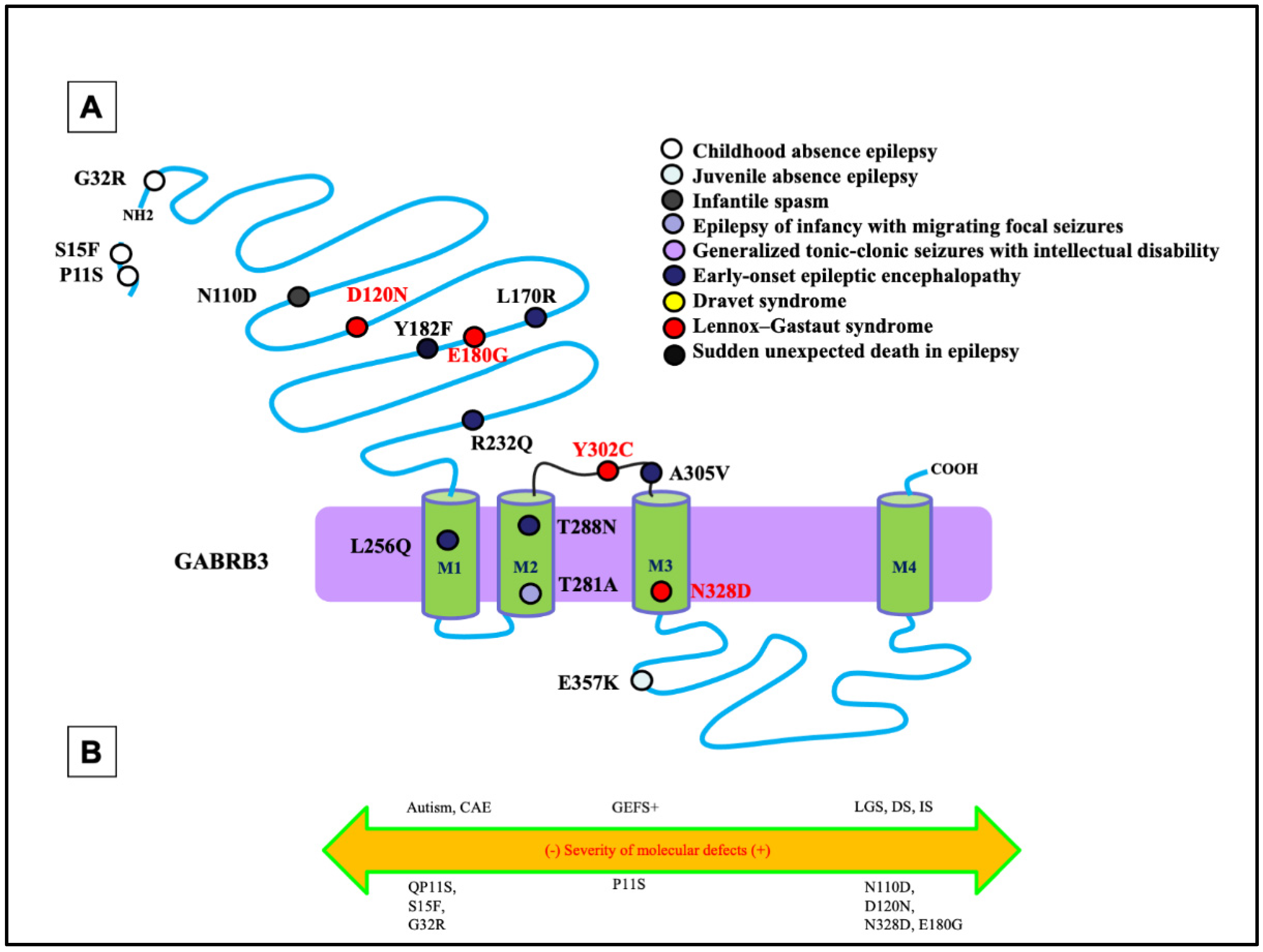
Figure 2. Epilepsy phenotype associated GABRB3 mutations. (A) Two-dimensional protein topology of the β3 subunit showing relative locations of mutations in human GABRB3 associated with various epilepsy syndromes and neurodevelopmental disorders. Each mutation is color coded to indicate associated diagnosis, and each dot represents the β3 subunit protein peptide. (B) Diagram dictating the severity of each mutation on a continuum of epilepsy phenotypes. The left side of the diagram infers a less severe epileptic phenotype, the middle dictates a moderate, and the right side of the diagram infers a more severe epileptic phenotype.
Mutations in GABRB3 have been frequently related to DEEs (Figure 2). A previous study evaluated the mutations identified by the Epilepsy Phenome/Genome Project (D120N, E180G, and Y302C) [7]. Follow-up functional assays identified the alterations of GABA-activated channel function, including reduced GABA-evoked current amplitudes, slowed activation, and accelerated deactivation [12]. This was inferred to be due to a reduction in the GABA potency, representing the concentration of GABA, and efficacy, representing the potential of GABA to bind, caused by the mutations. Specifically, the D120N mutation reduced GABA potency, whereas the E180G and Y302C mutations reduced GABA efficacy [12]. This implicates that different mutations can have separate effects on the inhibitory neurotransmission mechanisms. It is important to note that each mutation was in either loop A or loop B of the GABA-binding pocket or the M2-M3 loop, which is involved in the ligand-binding channel, gating-coupling mechanism. The specific pathomechanism could be the likely cause of the mutations disrupting coupling of GABA binding to channel gating.
The differential pathomechanisms in GABRB3 mutations associated with epilepsy with variable severities have been identified [13]. There was a thorough comparison of the GABRB3 (N328D) mutation associated with LGS with GABRB3 (E357K), a mutation associated with a less severe phenotype: juvenile absence epilepsy [13]. In the patient carrying the N328D mutation, there was presence of generalized tonic-clonic seizures and myoclonic seizures. The initial EEG examination showed ictal 2–2.5 Hz generalized polyspike-wave discharges during myoclonic seizures, interictal spike, and slow wave complexes, and irregular fast rhythms with slow waves were recorded during wakefulness [13]. It was identified that the LGS-associated GABRB3 (N328D) mutation caused more reduced cell-surface expression and synaptic presentation of α1β3γ2 than GABRB3(E357K) mutation associated with the juvenile absence epilepsy. However, both mutations caused trafficking defects due to endoplasmic reticulum retention of the mutant protein. Through use of a high-throughput flow cytometry assay, the surface expression of β3 and γ2 subunits for several GABRB3 mutations was evaluated. There was a significant reduction in the expression of the β3 and γ2 subunits at the cell surface for both mutations [13]. This implies that a mutation of this subunit not only prevents proper trafficking of the individual subunit itself but for the partnering wildtype subunits such as γ2 and α1 subunit expression, suggesting loss of whole receptor function instead of subunit alone. By comparing mutations in GABRB3 associated with different phenotypes or different mutations with the same phenotype such as LGS, it is likely that different mutations can have separate effects on the inhibitory neurotransmission mechanisms.
The impact of the GABRB3 mutation associated with LGS has also been investigated in mutant, knock-in Gabrb3+/D120N mice [9]. Like seizures observed in patients carrying the mutation, multiple types of spontaneous seizures including absence, myoclonic, tonic, and generalized tonic-clonic seizures were observed in mice of approximately four months of age. More specifically, the mutant mice showed a seizure frequency of 445 absence, 99 myoclonic, and 4 tonic seizures [9]. As there is a neuropsychiatric comorbidity in LGS, the disease-relevant behavioral abnormalities commonly observed in LGS such as ID and ASD have been evaluated with a battery of behavioral tests conducted in Gabrb3+/D120N mice. The Gabrb3+/D120N mice displayed impaired sociability and cognition evaluated with the three-chamber socialization test. In the Barnes maze test, the Gabrb3+/D120N mice showed a longer latency to find the target hole and increased number of errors in the learning trials. In the memory trial, there was also a significant increase in the number of errors to find the target hole, which both suggests learning and memory deficits in the mice [9].
References
- Kang, J.-Q. The GABAA Receptor 2 Subunit R43Q Mutation Linked to Childhood Absence Epilepsy and Febrile Seizures Causes Retention of 1 2 2S Receptors in the Endoplasmic Reticulum. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 8672–8677.
- Huang, X.; Hernandez, C.C.; Hu, N.; Macdonald, R.L. Three epilepsy-associated GABRG2 missense mutations at the γ+/β− interface disrupt GABAA receptor assembly and trafficking by similar mechanisms but to different extents. Neurobiol. Dis. 2014, 68, 167–179.
- Kang, J.-Q.; Shen, W.; Zhou, C.; Xu, D.; Macdonald, R.L. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat. Neurosci. 2015, 18, 988–996.
- Kang, J.-Q.; Shen, W.; Lee, M.; Gallagher, M.J.; Macdonald, R.L. Slow Degradation and Aggregation In Vitro of Mutant GABAA Receptor 2(Q351X) Subunits Associated with Epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13895–13905.
- Kang, J.-Q.; Shen, W.; Macdonald, R.L. The GABRG2 Mutation, Q351X, Associated with Generalized Epilepsy with Febrile Seizures Plus, Has Both Loss of Function and Dominant-Negative Suppression. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2845–2856.
- Warner, T.A.; Shen, W.; Huang, X.; Liu, Z.; Macdonald, R.L.; Kang, J.-Q. Differential molecular and behavioural alterations in mouse models of GABRG2haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum. Mol. Genet. 2016, 25, 3192–3207.
- Epi4K Consortium, Epilepsy Phenome/Genome Project; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221.
- Adak, P.; Sinha, S.; Banerjee, N. An Association Study of Gamma-Aminobutyric Acid Type A Receptor Variants and Susceptibility to Autism Spectrum Disorders. J. Autism Dev. Disord. 2021, 51, 4043–4053.
- Qu, S.; Catron, M.; Zhou, C.; Janve, V.; Shen, W.; Howe, R.K.; Macdonald, R.L. GABAA receptor β3 subunit mutation D120N causes Lennox–Gastaut syndrome in knock-in mice. Brain Commun. 2020, 2, fcaa028.
- Gurba, K.N.; Hernandez, C.; Hu, N.; Macdonald, R.L. GABRB3 Mutation, G32R, Associated with Childhood Absence Epilepsy Alters α1β3γ2L γ-Aminobutyric Acid Type A (GABAA) Receptor Expression and Channel Gating. J. Biol. Chem. 2012, 287, 12083–12097.
- Feucht, M.; Fuchs, K.; Pichlbauer, E.; Hornik, K.; Scharfetter, J.; Goessler, R.; Füreder, T.; Cvetkovic, N.; Sieghart, W.; Kasper, S.; et al. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol. Psychiatry 1999, 46, 997–1002.
- Ms, V.S.J.; Hernandez, C.C.; Bs, K.M.V.; Hu, N.; Macdonald, R.L. Epileptic encephalopathy de novoGABRBmutations impair γ-aminobutyric acid type A receptor function. Ann. Neurol. 2016, 79, 806–825.
- Shi, Y.-W.; Zhang, Q.; Cai, K.; Poliquin, S.; Shen, W.; Winters, N.; Yi, Y.-H.; Wang, J.; Hu, N.; Macdonald, R.L.; et al. Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 2019, 142, 3028–3044.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
4 times
(View History)
Update Date:
15 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No