Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yu Wang | -- | 1856 | 2022-09-13 08:49:29 | | | |
| 2 | Conner Chen | Meta information modification | 1856 | 2022-09-14 08:35:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yang, T.; Liu, D.; Fang, S.; Ma, W.; Wang, Y. Cytomegalovirus and Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/27114 (accessed on 28 June 2026).
Yang T, Liu D, Fang S, Ma W, Wang Y. Cytomegalovirus and Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/27114. Accessed June 28, 2026.
Yang, Tianrui, Delin Liu, Shiyuan Fang, Wenbin Ma, Yu Wang. "Cytomegalovirus and Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/27114 (accessed June 28, 2026).
Yang, T., Liu, D., Fang, S., Ma, W., & Wang, Y. (2022, September 13). Cytomegalovirus and Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/27114
Yang, Tianrui, et al. "Cytomegalovirus and Glioblastoma." Encyclopedia. Web. 13 September, 2022.
Copy Citation
Glioblastoma is the most common and aggressive malignancy in the adult central nervous system. Cytomegalovirus (CMV) plays a crucial role in the pathogenesis and treatment of glioblastoma.
cytomegalovirus
glioblastoma
carcinogenesis
1. Introduction
Glioblastoma (GBM) is the most common and aggressive tumor in the adult central nervous system. GBM has a dreadful prognosis, despite the combination of surgery, radiotherapy, and chemotherapy used to treat it. The median overall survival (OS) of GBM is approximately 14.4 months, and the 5-year survival rate is below 10% [1][2][3]. GBM represents one of the greatest challenges in the modern era, with a high recurrence rate and a low chance of survival. Therapeutic options are extremely limited at the first diagnosis and relapse. Recent studies have discovered the latent connection between GBM and cytomegalovirus (CMV), shedding light on the possibility of treating GBM with CMV-targeting therapy.
CMV is a double-stranded DNA virus and one of the largest and most complicated kinds of human herpes virus (HHV) [4]. CMV IgG antibodies are found in approximately 60% of adults in developed countries and 100% in developing countries [5]. Notably, the high CMV detection rate does not equate to high CMV activity. Most CMV infections are asymptomatic, establishing a lifelong latent infection. Only in immunosuppressed patients, such as patients with AIDS, organ transplantation patients, and infants [6], will CMV form inclusions in the cell nuclei, perinuclear spaces, and plasma, resulting in cell swelling (cytomegaly) and subsequent CMV disease [5]. CMV infection typically undergoes a three-phase process [7]: the immediate early phase (IE, 0 to 4 h after cell infection), delayed early phase (DE, 4 to 24 h after cell infection), and late phase (>24 h after cell infection). In the IE phase, the proteins IE-72 and IE-86 are expressed to alter the host cell environment and initiate DNA duplication. In the DE phase, non-structural proteins facilitate the viral DNA duplication and adjust the immune responses. Structural proteins, such as the envelope proteins pp65 and gB, are expressed in the late phase and aid in the virus assembly. Proteins, including IE, pp65, and gB, can serve as detection targets for CMV infection.
2. Detection of CMV
Currently, there are various methods for detecting CMV infection, including viral DNA detection, antigen and/or serum antibody testing, histopathological evidence, and viral culture. The most widely used indicator of CMV is pp65, which semi-quantitatively reflects the infection activity [8]. Serological tests for IgM and IgG antibodies are less quantitative and require a cut-off value with a higher specificity [9]. Pathology, immunohistochemistry (IHC), in situ hybridization (ISH), polymerase chain reaction (PCR), and tissue microarray (TMA) can also detect the CMV markers [10][11].
Sensitivity varies among different detection methods. A meta-analysis summarized the sensitivity levels of different methods and targets [12]. In CMV-positive patients, the IE protein had the highest association with CMV infection (odds ratio (OR) = 140), followed by the pp65 nucleic acid (OR = 18), pp65 protein (OR = 3.1), and gB nucleic acid (OR = 3.1). With respect to the detection methods, pathology analyses were considered to have high sensitivity. ISH had the highest correlation (OR = 28), polymerase chain reaction (PCR) had an OR of 3.7, and IHC had an OR of 3.5 [12]. The discrepancy between blood serum and whole blood samples should also be noted. The CMV DNA level detected was significantly higher in whole blood samples than that in serum [13]. The establishment of a low-grade infection in glioma cells also provides an explanation as to why CMV was not consistently detected by different groups [14].
3. Is CMV Infection Associated with GBM?
CMV has a high detection rate in many primary or metastatic tumors. For instance, in prostate, breast, and colorectal cancer, CMV infection rates are up to 90–100% [15][16][17]. In studies that examined glioma, medulloblastoma, meningioma, and neuroblastoma, CMV pp65 was positive across all tumor types regardless of age, sex, and WHO classification [18][19].
In glioma patients, the CMV detection rate is controversial. Studies have shown that the CMV detection rate is higher in glioma patients than in non-glioma patients. Previous research has suggested that the positive rate of CMV differed more extensively between GBM patients and control patients than those of Epstein Barr virus, human herpesvirus, and herpes simplex virus [20]. CMV DNA is detectable in most tumor samples and blood samples, with a higher detection rate than serum antibodies [11][21][22]. The detection of the CMV genome also confirmed the higher CMV detection rate in GBM patients than in brain tumor or epilepsy patients [23]. With respect to the histopathological evidence, the detection rates of CMV in non-tumor encephalopathies and normal brain samples were significantly lower than those in glioma samples [24]. However, other studies demonstrated that the CMV detection rate among the experimental group was not significantly different from that of the control group [25][26][27]. A meta-analysis summarized the findings of 32 independent studies with over 2000 patients and reported that the CMV detection rate varied extensively due to differences between populations and detection methods [12]. The overall CMV-positive rate in glioma patients was 63% and was significantly associated with gliomas (adjusted OR = 3). The detection rate was not significantly different between different glioma grades [12]. There is no final conclusion about the frequency of CMV in glioma.
4. CMV Infection Is Associated with the Prognosis of GBM
It has been reported that a higher expression of the IE protein was correlated with more aggressive tumor progression and shorter OS in extracranial tumors [17]. In GBM patients, positive serological and pathological CMV detection was associated with poor prognosis [28][29], and mainstream scholars agree that patients with higher IE protein expression had significantly shorter OS [24]. A meta-analysis that included seven studies identified no association between the CMV infection and prognosis [30], but among glioma patients with confirmed CMV infection, a low pathological positive rate was associated with better prognosis and longer survival [31][32]. With an IHC cut-off value of 25%, patients with a lower CMV-positive rate had a 20-month longer OS and an 8-month longer PFS than patients with a higher rate. The less positive subgroup also had a significantly higher 2-year survival rate (63.6% vs. 17.2%). However, the difference in median time to progression was non-significant between the lower and higher positive rate patients (for CMV-IEA: 14 vs. 6 months, and for CMV-LA: 8.25 vs. 5 months) [31]. Another case–control study investigated the recursive partition analysis (RPA) subclass, age, surgery, and adjuvant treatments among patients who survived over 18 months, of whom 40% had a low CMV-positive rate, and these patients survived for a median of 42.5 months, indicating that a low-grade CMV infection was strongly associated with long-term survival in GBM patients [32]. Comparatively, among patients with an OS shorter than 18 months, only 8% had a low positive rate, and 47.5% patients had more than 75% of their tumor cells infected [32]. Molecular analyses in CMV-positive rate subgroups have shown that the expression of CMV-IE was significantly associated with p53 mutations, telomerase activity, and several proto-oncogenes, resulting in a more aggressive tumor phenotype [31][33][34][35]. This observation further supports the hypothesis that CMV plays a pathogenetic role in GBM tumors, rather than representing an epiphenomenon, presenting a reasonable explanation for its poor prognosis.
5. Mechanisms of CMV-Related Glioma Tumorigenesis
Many preliminary studies have focused on the correlations between CMV and glioma and its tumorigenesis mechanisms from various perspectives. To date, theories regarding tumorigenesis include oncomodulation, oncogenic features, the regulation of the tumor microenvironment, regulation of epithelial-mesenchymal transition (EMT), and the overall regulation of the immune system (Figure 1).
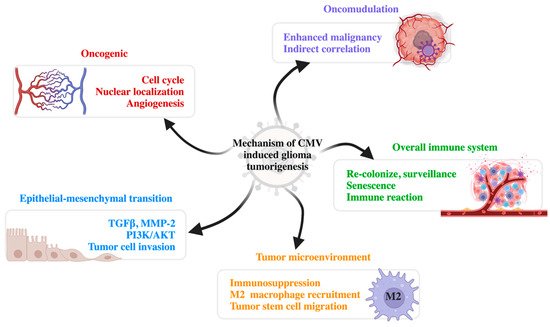
Figure 1. Mechanisms of CMV-induced glioma tumorigenesis. There are five major mechanisms of CMV tumorigenesis: oncogenic, oncomodulation, epithelial-mesenchymal transition, modified tumor microenvironment, and influence on the overall immune system. CMV, cytomegalovirus; TGFβ, transforming growth factor β; MMP-2, matrix metalloproteinase-2; PI3K/AKT, phosphatidylinositol 3-kinase/protein kinase B (PKB).
5.1. Oncomodulation
Although earlier studies reported that CMV had oncogenic abilities in in vitro experiments and experiments on immunodeficient mice [36], CMV was not able to transform normal human cells. Thus, CMV was considered to have an indirect influence on oncogenesis. Multiple studies have indicated that CMV can interfere with the cell cycle, induce telomerase activation and the DNA damage response, and thus inhibit apoptosis. For instance, the replication stress instigated by CMV infection stimulates the human DNA damage response. Meanwhile, viruses hijack the human stress response transcriptional factors to enhance their own expression of IE72 and IE86. In clinical testing, CMV protein markers are elevated after DNA damage by radiochemotherapy or the recurrence of GBM [37]. CMV is also capable of inducing angiogenesis and cell migration. CMV inhibits cell differentiation and facilitates cancer stem cell preservation. CMV can also induce the expression of oncogenes and inhibit tumor suppressors, such as the p53 mutation. The epigenetic regulation of cell proliferation and immune evasion was also observed in CMV infection [38].
5.2. Oncogenic Features
Researchers have discovered that there is a morphological transforming region II imbedded in the CMV genome, which can transform mouse fibroblast cells into malignant cells [39]. The IE protein expressed in the early phase of CMV infection can induce cell transformation through a ‘hit and run’ mechanism, which can activate telomerase to facilitate oncogenesis [40]. Additionally, the IE protein facilitates the correct nuclear localization of the CMV genome during mitosis through the chromatin-tethering domain. The IE protein can also maintain the mitotic cell cycle of the host cells and induce cell proliferation [41]. In addition, CMV expresses the US28 protein, which induces IL-6 expression and STAT3 phosphorylation, both promoting paracrine signaling in oncogenesis [42]. A higher level of VEGF expression is also induced by CMV, which recruits the perivascular cells and promotes angiogenesis [43]. Other studies have indicated that CMV is associated with GSK-3β inhibition and the activation of the WNT, NF-κB, EGFR, ERK, amphiregulin, and SOX-2 pathways [44][45].
5.3. Tumor Microenvironment
CMV infection induces COX-2 and 5-LO expression in the tumor microenvironment, leading to the expression of PGE2 and leukotriene as part of inflammatory processes. PGE2 induces cell proliferation and angiogenesis, inhibits apoptosis, and activates invasion, encouraging the formation of the tumor microenvironment [46][47]. CMV infection of the monocytes and neural stem cells generates IL-10 and induces the recruitment of the tumor microenvironment-associated monocytes (macrophages and microglial cells). These monocytes present with the M2 immunosuppressive phenotype, with down-regulated MHC and costimulatory molecules. CMV infection also upregulates B7-H1, an immunosuppressive molecule, which enhances tumor stem cell migration [48].
5.4. Epithelial–Mesenchymal Transition
Epithelial–mesenchymal transition (EMT) is an important mechanism of epithelial tumor metastasis. In addition, TGFβ is the lynchpin molecule of the EMT mechanism. CMV induces the expression of TGFβ through multiple pathways [49]. For instance, the IE-72 and IE-86 proteins activate extracellular latent TGFβ1 through matrix metalloproteinase-2 (MMP-2) [49][50]. US28 also facilitates EMT by inducing GSK3β activity. The phosphorylation of GSK3β activates the transcription factors of oncogenes, such as Smads and Snail, to trigger EMT [39]. Additionally, by regulating the IL-10, COX-2, RAS/ERK, and PI3K/AKT pathways, CMV can promote the invasion of tumor cells [51].
5.5. Overall Immune System
CMV can also influence the mode of overall immunity. This kind of immune modulation might be related to patient age [52]. In young patients, CMV can generate a broad T-cell receptor pool, enhance the range and ability of T-cell recognition, and enhance anti-tumor immune reactions. The upregulation of CX3CR1 indicates that CMV-specific T cells can recolonize and establish T-cell surveillance. However, in elderly patients, CMV primarily results in T-cell senescence and, therefore, a low survival rate. Unlike PD-1, which can be reversed by checkpoint inhibitors, this senescence is irreversible. Senescent T cells can still access the memory T-cell pool and compete with the binding of specific T-cell receptors, altering the immune reaction and suppressing the overall anti-tumor immune response [53][54].
References
- Jiang, T.; Nam, D.-H.; Ram, Z.; Poon, W.-S.; Wang, J.; Boldbaatar, D.; Mao, Y.; Ma, W.; Mao, Q.; You, Y.; et al. Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2020, 499, 60–72.
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22 (Suppl. S2), iv1–iv96.
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466.
- Landolfo, S.; Gariglio, M.; Gribaudo, G.; Lembo, D. The human cytomegalovirus. Pharmacol. Ther. 2003, 98, 269–297.
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297.
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102.
- Picarda, G.; Benedict, C.A. Cytomegalovirus: Shape-Shifting the Immune System. J. Immunol. 2018, 200, 3881–3889.
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplant recipients-Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512.
- Halwachs-Baumann, G. Recent developments in human cytomegalovirus diagnosis. Expert. Rev. Anti. Infect. Ther. 2007, 5, 427–439.
- Cobbs, C.S. Cytomegalovirus and brain tumor: Epidemiology, biology and therapeutic aspects. Curr. Opin. Oncol. 2013, 25, 682–688.
- Charles, S.; Cobbs, L.H.; Minu Samanta, G.; Gillespie, Y.; Bharara, S.; Peter, H.; King, L.; Burt Nabors, C.; Cobbs, G.; William, J.B. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res. 2002, 62, 3347–3350.
- Farias, K.; Moreli, M.L.; Floriano, V.G.; da Costa, V.G. Evidence based on a meta-analysis of human cytomegalovirus infection in glioma. Arch. Virol. 2019, 164, 1249–1257.
- Kullberg-Lindh, C.; Olofsson, S.; Brune, M.; Lindh, M. Comparison of serum and whole blood levels of cytomegalovirus and Epstein-Barr virus DNA. Transpl. Infect. Dis. 2008, 10, 308–315.
- Michaelis, M.; Doerr, H.W.; Cinatl, J., Jr. Oncomodulation by human cytomegalovirus: Evidence becomes stronger. Med. Microbiol. Immunol. 2009, 198, 79–81.
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; I Bland, K.; Cobbs, C.S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563.
- Samanta, M.H.L.; Klemm, K.; Britt, W.J.; Cobbs, C.S. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J. Urol. 2003, 170, 998–1002.
- Taher, C.; Frisk, G.; Fuentes, S.; Religa, P.; Costa, H.; Assinger, A.; Vetvik, K.K.; Bukholm, I.R.; Yaiw, K.-C.; Smedby, K.E.; et al. High Prevalence of Human Cytomegalovirus in Brain Metastases of Patients with Primary Breast and Colorectal Cancers. Transl. Oncol. 2014, 7, 732–740.
- Libard, S.; Popova, S.N.; Amini, R.-M.; Kärjä, V.; Pietiläinen, T.; Hämäläinen, K.M.; Sundström, C.; Hesselager, G.; Bergqvist, M.; Ekman, S.; et al. Human Cytomegalovirus Tegument Protein pp65 Is Detected in All Intra- and Extra-Axial Brain Tumours Independent of the Tumour Type or Grade. PLoS ONE 2014, 9, e108861.
- Wolmer-Solberg, N.; Baryawno, N.; Rahbar, A.; Fuchs, D.; Odeberg, J.; Taher, C.; Wilhelmi, V.; Milosevic, J.; Mohammad, A.A.; Martinsson, T.; et al. Frequent detection of human cytomegalovirus in neuroblastoma: A novel therapeutic target? Int. J. Cancer 2013, 133, 2351–2361.
- Wrensch, M.; Weinberg, A.; Wiencke, J.; Miike, R.; Barger, G.; Kelsey, K. Prevalence of Antibodies to Four Herpesviruses among Adults with Glioma and Controls. Am. J. Epidemiol. 2001, 154, 161–165.
- Rahbar, A.; Peredo, I.; Solberg, N.W.; Taher, C.; Dzabic, M.; Xu, X.; Skarman, P.; Fornara, O.; Tammik, C.; Yaiw, K.; et al. Discordant humoral and cellular immune responses to Cytomegalovirus (CMV) in glioblastoma patients whose tumors are positive for CMV. Oncoimmunology 2015, 4, e982391.
- Söderberg-Nauclér, C.; Stragliotto, G. Valganciclovir in Patients with Glioblastoma. N. Engl. J. Med. 2013, 369, 2064–2066.
- Ranganathan, P.; Clark, P.A.; Kuo, J.S.; Salamat, M.S.; Kalejta, R.F. Significant association of multiple human cytomegalovirus genomic Loci with glioblastoma multiforme samples. J. Virol. 2012, 86, 854–864.
- Wen, L.; Zhao, F.; Qiu, Y.; Cheng, S.; Sun, J.Y.; Fang, W.; Rayner, S.; McVoy, M.A.; Jiang, X.J.; Tang, Q.; et al. Human cytomegalovirus DNA and immediate early protein 1/2 are highly associated with glioma and prognosis. Protein Cell 2020, 11, 525–533.
- Priel, E.; Wohl, A.; Teperberg, M.; Nass, D.; Cohen, Z.R. Human cytomegalovirus viral load in tumor and peripheral blood samples of patients with malignant gliomas. J. Clin. Neurosci. 2015, 22, 326–330.
- Solomon, I.H.; Ramkissoon, S.H.; Milner, D.A., Jr.; Folkerth, R.D. Cytomegalovirus and Glioblastoma: A Review of Evidence for Their Association and Indications for Testing and Treatment. J. Neuropathol. Exp. Neurol. 2014, 73, 994–998.
- Ghaffari, H.; Tavakoli, A.; Faranoush, M.; Naderi, A.; Kiani, S.J.; Sadeghipour, A.; Javanmard, D.; Farahmand, M.; Ghorbani, S.; Sedaghati, F.; et al. Molecular Investigation of Human Cytomegalovirus and Epstein-Barr virus in Glioblastoma Brain Tumor: A Case-Control Study in Iran. Iran. Biomed. J. 2021, 25, 426–433.
- Foster, H.; Piper, K.; Depledge, L.; Li, H.-F.; Scanlan, J.; Jae-Guen, Y.; Boeckh, M.; Cobbs, C.; Yoon, J.-G. Human cytomegalovirus seropositivity is associated with decreased survival in glioblastoma patients. Neuro-Oncol. Adv. 2019, 1, vdz020.
- Lisyany, N.I.; Klyuchnikova, A.A.; Belskaya, L.N.; Lisyany, A.A.; Gnedkova, I.A. Cytomegaloviruses and malignant brain tumors. Exp. Oncol. 2019, 41, 300–303.
- Cai, Z.; Yang, S.; Li, X.; Chen, F.; Li, W. Viral infection and glioma: A meta-analysis of prognosis. BMC Cancer 2020, 20, 549.
- Rahbar, A.; Orrego, A.; Peredo, I.; Dzabic, M.; Wolmer-Solberg, N.; Strååt, K.; Stragliotto, G.; Söderberg-Nauclér, C. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J. Clin. Virol. 2013, 57, 36–42.
- Rahbar, A.; Stragliotto, G.; Orrego, A.; Peredo, I.; Taher, C.; Willems, J.; Söderberg-Naucler, C. Low levels of Human Cytomegalovirus Infection in Glioblastoma multiforme associates with patient survival—A case-control study. Herpesviridae 2012, 3, 3.
- Strååt, K.; Liu, C.; Rahbar, A.; Zhu, Q.; Liu, L.; Wolmer-Solberg, N.; Lou, F.; Liu, Z.; Shen, J.; Jia, J.; et al. Activation of Telomerase by Human Cytomegalovirus. J. Natl. Cancer Inst. 2009, 101, 488–497.
- Cinatl, J., Jr.; Nevels, M.; Paulus, C.; Michaelis, M. Activation of telomerase in glioma cells by human cytomegalovirus: Another piece of the puzzle. J. Natl. Cancer Inst. 2009, 101, 441–443.
- Maleki, F.; Sadigh, Z.A.; Sadeghi, F.; Muhammadnejad, A.; Farahmand, M.; Parvin, M.; Shirkoohi, R. Human cytomegalovirus infection in Iranian glioma patients correlates with aging and tumor aggressiveness. J. Med. Virol. 2020, 92, 1266–1276.
- Geder, L.S.E.; Rohner, T.J.; Rapp, F. Cytomegalovirus and cancer of the prostate: In vitro transformation of human cells. Cancer Treat. Rep. 1977, 61, 139–146.
- Merchut-Maya, J.M.; Bartek, J., Jr.; Bartkova, J.; Galanos, P.; Pantalone, M.R.; Lee, M.; Cui, H.L.; Shilling, P.J.; Brøchner, C.B.; Broholm, H.; et al. Human cytomegalovirus hijacks host stress response fueling replication stress and genome instability. Cell Death Differ. 2022, 29, 1639–1653.
- Joseph, G.P.; McDermott, R.; Baryshnikova, M.A.; Cobbs, C.S.; Ulasov, I.V. Cytomegalovirus as an oncomodulatory agent in the progression of glioma. Cancer Lett. 2017, 384, 79–85.
- Soderberg-Naucler, C.; Johnsen, J.I. Cytomegalovirus in human brain tumors: Role in pathogenesis and potential treatment options. World J. Exp. Med. 2015, 5, 1–10.
- Shen, Y.; Zhu, H.; Shenk, T. Human cytomagalovirus IE1 and IE2 proteins are mutagenic and mediate "hit-and-run" oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 3341–3345.
- Lyon, S.M.Y.K.; Paulus, C.; Nevels, M.; Kalejta, R.F. Human Cytomegalovirus Genomes Survive Mitosis via the IE19 Chromatin-Tethering Domain. mBio 2020, 29, e02410–e02420.
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Söderberg-Nauclér, C.; Smit, M.J. HCMV-Encoded Chemokine Receptor US28 Mediates Proliferative Signaling Through the IL-6–STAT3 Axis. Sci. Signal. 2010, 3, ra58.
- Krenzlin, H.; Behera, P.; Lorenz, V.; Passaro, C.; Zdioruk, M.; Nowicki, M.O.; Grauwet, K.; Zhang, H.; Skubal, M.; Ito, H.; et al. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J. Clin. Investig. 2019, 129, 1671–1683.
- Soroceanu, L.; Cobbs, C.S. Is HCMV a tumor promoter? Virus Res. 2011, 157, 193–203.
- Ulasov, I.V.; Kaverina, N.V.; Ghosh, D.; Baryshnikova, M.A.; Kadagidze, Z.G.; Karseladze, A.I.; Baryshnikov, A.Y.; Cobbs, C.S. CMV70-3P miRNA contributes to the CMV mediated glioma stemness and represents a target for glioma experimental therapy. Oncotarget 2017, 8, 25989–25999.
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; Dallas, S.R.M.; Smit, M.; Soroceanu, L.; Cobbs, C.S. The HCMV and Gliomas Symposium. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro-Oncology 2012, 14, 246–255.
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Walsum, M.S.-V.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The Human Cytomegalovirus–Encoded Chemokine Receptor US28 Promotes Angiogenesis and Tumor Formation via Cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869.
- Dziurzynski, K.; Wei, J.; Qiao, W.; Hatiboglu, M.A.; Kong, L.-Y.; Wu, A.; Wang, Y.; Cahill, D.; Levine, N.; Prabhu, S.; et al. Glioma-Associated Cytomegalovirus Mediates Subversion of the Monocyte Lineage to a Tumor Propagating Phenotype. Clin. Cancer Res. 2011, 17, 4642–4649.
- Zhu, X.; Hu, B.; Hu, M.; Qian, D.; Wang, B. Human cytomegalovirus infection enhances invasiveness and migration of glioblastoma cells by epithelial-to-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2020, 13, 2637–2647.
- Shimamura, M.; Murphy-Ullrich, J.E.; Britt, W.J. Human cytomegalovirus induces TGF-beta1 activation in renal tubular epithelial cells after epithelial-to-mesenchymal transition. PLoS Pathog. 2010, 6, e1001170.
- Cobbs, C. Cytomegalovirus is a tumor-associated virus: Armed and dangerous. Curr. Opin. Virol. 2019, 39, 49–59.
- Luo, X.-H.; Meng, Q.; Rao, M.; Liu, Z.; Paraschoudi, G.; Dodoo, E.; Maeurer, M. The impact of inflationary cytomegalovirus-specific memory T cells on anti-tumour immune responses in patients with cancer. Immunology 2018, 155, 294–308.
- Kallemeijn, M.J.; Boots, A.M.H.; Van Der Klift, M.Y.; Brouwer, E.; Abdulahad, W.H.; Verhaar, J.; Van Dongen, J.; Langerak, A.W. Ageing and latent CMV infection impact on maturation, differentiation and exhaustion profiles of T-cell receptor gammadelta T-cells. Sci. Rep. 2017, 7, 5509.
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
885
Revisions:
2 times
(View History)
Update Date:
14 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No