Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Meraviglia-Crivelli | -- | 3596 | 2022-09-08 11:26:41 | | | |
| 2 | Sirius Huang | Meta information modification | 3596 | 2022-09-09 03:00:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Meraviglia-Crivelli, D.; Zheleva, A.; Barainka, M.; Moreno, B.; Villanueva, H.; Pastor, F. Approaches to Enhance Tumor Antigenicity. Encyclopedia. Available online: https://encyclopedia.pub/entry/26998 (accessed on 26 June 2026).
Meraviglia-Crivelli D, Zheleva A, Barainka M, Moreno B, Villanueva H, Pastor F. Approaches to Enhance Tumor Antigenicity. Encyclopedia. Available at: https://encyclopedia.pub/entry/26998. Accessed June 26, 2026.
Meraviglia-Crivelli, Daniel, Angelina Zheleva, Martin Barainka, Beatriz Moreno, Helena Villanueva, Fernando Pastor. "Approaches to Enhance Tumor Antigenicity" Encyclopedia, https://encyclopedia.pub/entry/26998 (accessed June 26, 2026).
Meraviglia-Crivelli, D., Zheleva, A., Barainka, M., Moreno, B., Villanueva, H., & Pastor, F. (2022, September 08). Approaches to Enhance Tumor Antigenicity. In Encyclopedia. https://encyclopedia.pub/entry/26998
Meraviglia-Crivelli, Daniel, et al. "Approaches to Enhance Tumor Antigenicity." Encyclopedia. Web. 08 September, 2022.
Copy Citation
Cancer immunotherapy has revolutionized the oncology field, but many patients still do not respond to the immunotherapy approaches. One of the main challenges in broadening the range of responses to this type of treatment is the limited source of tumor neoantigens. New approaches must be taken into consideration to overcome these shortcomings. The possibility of making tumors more antigenic represents a promising front to further improve the success of immunotherapy in cancer.
neoantigens
tumor immunity
cancer immunotherapy
1. Introduction
Cancer is one of the main causes of death in developed countries, along with cardiovascular diseases [1]. Consequently, there is a continuous need to find novel strategies to fight cancer. Immunotherapy has revolutionized the field of cancer therapy in the last years as it has been shown to work in numerous successful clinical trials [2][3][4][5][6] for different types of tumors. Active cancer immunotherapy is aimed at eliciting an endogenous immune response to seek and destroy delectably malignant cells. The efficiency and selectivity of the immune response are basically driven by the presence of tumor antigens. While most current cancer immunotherapy approaches focus on tuning tumor-reactive T-cell functionality, there are still few strategies aimed at enhancing tumor antigenicity.
2. Alteration in Antigen Presentation Pathways to Elicit Tumor Antigens
CD8 T cells recognizing peptide antigens presented on the tumor cell surface are bound to MHC-I molecules. The main source of MHC-I peptides is obtained through the protein degradation that takes place in the proteasome, generally in a ubiquitin-dependent manner. After proteolysis, the resultant peptides are imported into the endoplasmic reticulum (ER) via the transporter TAP and finally loaded into the MHC-I that presents the antigens on the cell surface so that CD8 T cells can scan and recognize them (Figure 1). Given the importance of the ubiquitin pathway in the processing of MHC-I peptides, once a strong antigen has been characterized, and its source is known, it would be interesting to boost the degradation of the original protein in order to enhance the peptide presentation in cancer cells. This is what proteolysis targeting chimeras (PROTACs) do. PROTACs are small molecules that target a specific protein to proteasomal degradation by recruiting ubiquitin E3 ligases to knock down protein activity for therapeutic purposes. In order to achieve it, PROTACs have two functional cores, one acting as a ligand of the target protein and another recruiting the ubiquitin E3 ligases. Jensen and colleagues [7] tested three different PROTACs and were able to track the resultant peptides in the immunopeptidome of the treated cells. Massafra and colleagues [8], on the other hand, demonstrated that the peptide presentation induced with PROTAC treatment could activate T cells against human cancer cell lines in vitro. These types of drugs could be used to elicit an immune response against common tumor antigens (e.g., tumor oncogenes).
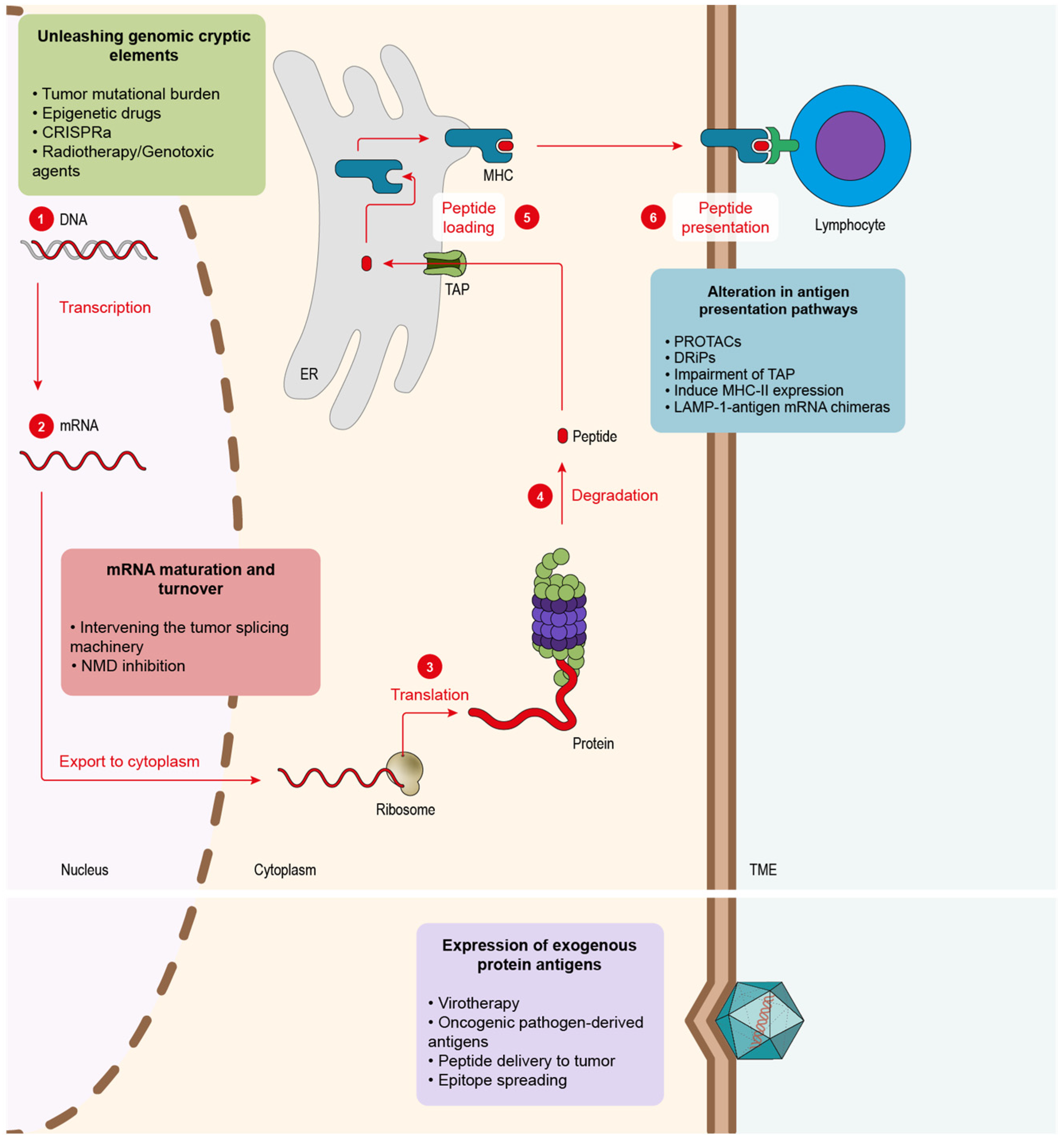
Figure 1. Tumor antigenicity can be enhanced at different levels. Endogenous tumor antigens can be fostered by altering different pathways in the tumor cells: 1. Genomic DNA contains an excellent source of dormant antigens that can be unleashed via several strategies: epigenetic drugs, radiotherapy, genotoxic drugs or CRISPRa. 2. Modulation of mRNAs maturation and homeostasis can lead to novel tumor antigens. Aberrant mRNAs might be generated via splicing inhibition or NMD blockade. 3–6. Proteins must be degraded to obtain antigenic peptides that are refined in the ER until the final ones are presented via MHC. Several strategies can be used to boost peptide-derived neoantigens at the protein level: PROTACs that induce selective target protein degradation, approaches that favor DRiP generation, TAP inhibition, induction of MHC-II presentation in tumor cells. Another source of antigens can be supplied by exogenous proteins.
The key transporter of peptides into the ER is TAP, a heterodimer formed by TAP1 and TAP2 subunits. The inhibition of this transporter partially inhibits the canonical peptide presentation, thus favoring an alternative source of peptides [9]. Hence, the impairment of TAP function in tumor cells offers two main advantages: (1) it triggers the presentation of peptides by the non-canonical pathway and thus makes them highly antigenic since they are not present in normal cells [10]; (2) these non-TAP dependent antigens might be mutation-independent, which makes them more prone to be shared across all tumor types [10][11]. In order to inhibit TAP, specifically in cancer cells, Garrido and colleagues [12] designed an aptamer-siRNA chimera that was able to target the malignant cells and trigger the impairment of TAP. Their results showed an increase in T-cell infiltration followed by a decrease in tumor growth in mice and better overall survival. Finally, TAP inhibition presented an additive effect with ICB therapy (anti-PD-1 antibody).
Apart from the MHC-I-dependent peptides, which are recognized by CD8 T cells, APCs also count, with MHC-II being recognized by CD4 helper lymphocytes. CD4 T lymphocytes play an important role in assisting the antitumor immune response [13]. MHC-II expression in tumor cells was reported in different cancer types, such as melanoma [14], in which it correlated with better clinical outcomes and response to PD-1/PD-L1 ICB therapy or breast cancer [15], and is also associated with better prognosis and higher immune infiltration. Some studies performed in mice exploring the induction of MHC-II expression in cancer cells presented promising results in breast cancer models [16].
Another way to boost MHC-II-dependent antigen presentation consists in delivering the antigen through the lysosomal pathway, such as the lysosome-associated membrane proteins (LAMPs). In order to achieve this, LAMP-1 and the antigen’s mRNA are fused to generate a chimeric mRNA that is then transfected into DCs. This approach was tested with carcinoembryonic antigen (CEA). Researchers observed the expansion of CEA-specific CD4+ T cells, which led to a stronger CD8 T-cell cytotoxic response to the same antigen in vitro [17]. The concept was tested in different phase-I clinical trials in glioblastoma patients (NCT02529072, NCT00626483, NCT00639639), showing an improvement in the overall survival of patients. In addition, the treatment is ongoing through several phase-II clinical trials trying to improve efficacy via combination with other drugs, such as antagonistic antibodies, GM-CSFG or tetanus toxoid preconditioning to enhance DC migration (NCT02366728, NCT03927222, NCT03688178, NCT02465268).
It has been discussed the peptides originated from the degradation of full proteins and how they can be tuned to enhance antigen presentation. However, another source that would likely lead to fast-presented peptides is defective ribosomal products (DRiPs). The DRiP hypothesis suggests that peptides could emerge from translation products that cannot, or do not, achieve a stable structure and are rapidly degraded [18][19]. The existence of DRiPs was studied mostly in viruses. Some studies proved that MHC-I peptides derived from viral products could be detected before the expression of the viral proteins per se [20][21]. In tumors, a work from Yewdell’s group showed that the knockdown of certain ribosomal proteins could boost the externalization of some MHC-I haplotypes without changing the total expression of MHC-I in the cell. This study also points to the fact that targeting ribosomes in cancer cells to change the antigen repertoire might help the immune system detect them as well as elicit strong antitumor responses [22].
3. Alteration of mRNA Maturation and Turnover to Foster New Tumor Antigens
Another strategy to induce the emergence of neoantigens is exploiting mRNA maturation and homeostasis (Figure 1). Errors during mRNA maturation may lead to aberrant transcripts that might, in turn, codify for potential neoantigens. Impaired expression of splicing modulators across several cancer types was detected, as in the case of U2AF1 and SF3B1, which were found mutated in myelodysplasia and chronic lymphocytic leukemia [23][24]. Additionally, a study performed on lung adenocarcinoma discovered that splicing impairment in cancer cells triggers the presentation of a new plethora of neoantigens derived from aberrant transcripts [25]. They also were able to identify the novel peptides and validate them in humanized mice. In order to exploit these splicing-originated neoantigens, some studies tested different antitumor therapies using splicing inhibitors. In 2021, Lu and colleagues [26] implemented two different splicing inhibitors, Indisulam and MS-023, showing that they could elicit a potent antitumor response mediated by neoantigens, which they identified while analyzing the immunopeptidome. They also demonstrated that peptides could be used to vaccinate mice and induce the activation of the immune system against the tumor cell in vitro. This strategy showed significant synergy in combination with PD-1 ICB therapy. Closer to the clinic is the splicing inhibitor E7820, which is currently in a phase-II trial (NCT05024994) in patients with myeloid cancers that show mutations in splicing factors. Apart from the neoantigen splicing inhibition awakening, the aberrant splicing machinery that many tumor cells harbor can offer further promising therapeutical opportunities that may complement the boost of neoantigen presentation. North and colleagues [27] showed that it was possible to deliver a well-characterized druggable target protein as an unprocessed mRNA that could be only spliced by the malignant cells. In this work, the authors employed cells that presented a mutated form of the splicing factor SF3B1. Cells were transduced with herpes simplex virus–thymidine kinase (HSV–TK) mRNA containing synthetic introns that could only be spliced by cells containing aberrant SF3B1, leaving healthy tissues untouched. In this way, only tumor cells were capable of processing the HSV-TK transcript, which conferred them sensibility to ganciclovir, an FDA-approved compound for herpes simplex [27].
Cancer cells can harbor multiple mutations in their genomes that result in the transcription of aberrant mRNAs. Nevertheless, not all mutations have the same immunogenic potential. In order to address this issue, the mutation type needs to be considered. According to its origin, we may find two kinds of mutation: (1) single-point mutations and (2) frameshift mutations. (1) Single-point mutations cause only one nucleotide substitution in the DNA, which leads, in the best-case scenario, to a new amino acid in the peptide sequence. (2) Frameshift mutations, on the other hand, trigger the formation of a novel sequence, opening the possibility of a new array of neoantigens radically different from the initial protein [28]. Despite their antigenic potential, many frameshift mutations lead to the creation of premature stop codons (PTCs). PTCs in messenger RNA (mRNA) are identified by the nonsense-mediated decay (NMD) machinery and degraded, so the potential antigens coded by this aberrant transcript are lost. It was observed that some PTC-containing mRNAs manage to escape NMD, supporting the importance of these NMD-dependent antigens in the clinic. In these cases, patients show a significantly better response to ICB therapy [29][30]. A strategy to recover the remaining peptides that NMD suppresses consists of compromising NMD activity. Pastor et al. (2010) [31] showed that NMD inhibition leads to tumor immunity by possible stabilization and presentation of this type of neoepitopes. In addition, recent work reported that NMD inhibition via an aptamer-siRNA chimera induced a boost in immune infiltrate and slowed tumor growth [32][33]. Another evidence of the importance of NMD in cancer was observed in mismatch repair (MMR) deficient colorectal cancer (CRC) with microsatellite instability (MSI). These types of tumors accumulate a high number of mutations due to their poor DNA repair capacity. It was observed that CRC MSI+ tumors display high NMD activity, probably to cope with the substantial levels of aberrant transcripts from the mutant genes. In fact, NMD inhibition was found to present a deleterious effect, slowing cell growth in vitro and tumor growth [34].
4. Unleashing Genomic Cryptic Elements
The principal sources of antigens at mRNA and their relevance in cancer immunotherapy have been reviewed. mRNAs, however, represent only a small fraction of the genome that is actively transcribed. Targeting the genome of cancer cells could trigger a vast array of novel antigenic peptides [35]. The main sources of neoantigens comprised in the genome originated from mutations present in malignant cells, which they acquire during the process of tumorigenesis (Figure 1). The presence of mutations, also known as TMB, was determined as a key biomarker in cancer. Clinical trials in several cancer types, such as lung [4] and melanoma [36], showed that high TMB correlated with better prognosis in combination with different ICB immunotherapies. One special case is tumors with microsatellite instability caused by a deficiency in MMR. In normal cells, MMR is a housekeeping mechanism that corrects base-to-base mismatches produced by exposure to DNA damage mediated by exogenous chemicals or physical agents (e.g., cigarette smoke) as well as some endogenous reactive metabolites (e.g., oxygen reactive species). In tumors that lack MMR, DNA accumulates a high number of somatic mutations, leading to the production of neoantigens associated with these mutations [37]. The presence of this potent immune response leads to boosting ICB treatment outcomes [6][38] in these tumors with MMR.
Genomes present multiple dormant elements, such as mobile elements, viruses and mutated gene copies [39] that could be able to originate different neoantigens. Epigenetic machinery executes vital regulatory processes that regulate the expression or repression of many elements in the cell genome [40]. Intervening in epigenetic events to activate genes involved in the MHC presentation pathway, immune-checkpoint genes such as PD-L1 (which can be targeted with ICB therapies or trigger the emergence of neoantigens) hold vast potential for the development of novel strategies to make tumors more antigenic. One of the epigenetic processes that can be targeted is DNA methylation, which is involved in gene repression and catalyzed by DNA methyltransferases (DNMTs). DNA methylation inhibitors, such as 5-aza-2-deoxycytidine (DAC), were FDA-approved for the treatment of hematological malignancies [41]. Preclinical data from this study showed that DAC might induce the demethylation of aberrant CpG islands producing dsRNAs that end up in the upregulation of type-III interferons (IFN). However, the mechanism of efficacy of this compound remains controversial. In addition to global demethylation, some studies point to DAC leading to the inhibition of NMD [42], which triggers the stabilization of aberrant mRNAs leading to the emergence of a potential array of neoantigens. Recently, a novel study showed that treatment with DAC in glioblastoma, a tumor with a really low TMB, induces the presentation of MHC-I-dependent neoantigens [43], demonstrating the high potential of epigenetic drugs in cancers with low mutational levels. The researchers treated patient-derived glioblastoma cells with DAC and co-cultured them with tumor-infiltrating lymphocytes (TILs) isolated from patients ex vivo, showing a significant immune reactivity boost when tumor cells were treated with DAC.
In addition, a phase-II clinical trial with guadecitabine, a decitabine analog, showed promising clinical results in patients with peripheral T-cell lymphoma. Guadecitabine upregulated proinflammatory signaling pathways (e.g., type I and II interferons, TNF-α and the JAK/STAT pathways). Moreover, genes associated with antigen presentation were also upregulated (e.g., β-2-microglobulin, TAP1) [44].
Similarly, another strategy to ‘awaken’ antigens consists in employing a variant of CRISPR/Cas technology called CRISPR activation (CRISPRa). CRISPRa comprises a catalytically dead Cas9 (dCas9) but keeps its ability to bind to genomic DNA via recognition of the small guide RNA (sgRNA) while engineered with transcription activators. Thus, after the docking of Cas9 to the target gene, its expression is triggered [45]. In the context of tumor antigenicity, Wang and colleagues showed that the activation of different genes through a multiplexed library of sgRNAs induced the presentation of antigens by cancer cells and elicited a potent antitumor immune response in mice [46].
Not all tumors present a high TMB that can be targeted to induce the presentation of neoantigens. Classic tumor therapies such as radiotherapy or chemotherapy drugs, such as cisplatin, present a strong genotoxic effect on DNA, causing lethal mutations that lead to cell death. Interestingly, the property of these treatments to induce mutations shows a clear potential in turning tumors more antigenic. There is some evidence of radiotherapy enhancing antitumor immune response by upregulating the expression of mutated genes leading to the presentation of neoantigens [47]. In this work, the authors managed to identify MHC-I and MHC-II-dependent peptides induced by radiotherapy treatment. Vaccination with a combination of these antigens, moreover, slowed tumor growth. In the clinic, radiotherapy has shown that it can dramatically improve the therapeutical outcome of ICB in metastatic cancers [48], which the authors hypothesize might be due to the antigen-presentation boost radiotherapy induces.
5. Expression of Exogenous Protein Antigens
mRNA homeostasis in cancer cells has proved to offer different strategies to promote tumor immunity, such as NMD or splicing modulation. Genomic mutations constitute a further important source of neoantigens that can trigger the immunogenicity of malignant cells. However, as reviewed here, not all tumors present a high TMB. Epigenetic drugs and splicing modulators might cause certain side effects such as short-term memory loss, thrombocytopenia, anorexia, fatigue, bleeding, anemia or joint pain [49][50]. A potential therapeutical approach to overcome this limitation is oncolytic viruses (Figure 1). Some strains present the ability to infect malignant cells preferentially [51], which makes them an interesting tool for developing therapies. Additionally, viruses can be modified in order to redirect their tropism towards cancer cells [51]. Another advantage of these pathogens is that they are detected as foreign elements by the immune system, which triggers strong immune responses [51]. Viral proteins and nucleic acids can serve as a source of neoantigens if presented specifically in tumor cells [52]. This is quite relevant for tumors that lack a high TMB and show low T-cell infiltration levels. Friedman and colleagues (2021) published a clinical trial demonstrating that the treatment with the oncolytic virus G207, engineered from the herpes simplex virus type 1 (HSV-1), which was capable of increasing the immune infiltrate of the tumors improving the patient’s survival [53]. Viruses were also widely engineered as vectors to express proinflammatory proteins [54]. Preclinical data from experiments performed in mice showed that the oncolytic virus Delta-24-RGD could be engineered to express costimulatory ligands, such as 4-1BBL or OX40L, in the surface of malignant cells, increasing tumor infiltration [55][56]. In this line of work, oncolytic viruses can be modified to express cytokines. The most successful approach to this kind of strategy is Talimogene laherparepvec (T-VEC), based on herpes simplex type-1 derived oncolytic virus, designed to replicate specifically in malignant cells and to express GM-CSF. During a clinical trial in advanced melanoma, T-VEC therapy showed a significant improvement in overall survival in the patients [57].
Similarly, a virus that does not show oncolytic properties can serve as a source of antigens. The influenza virus is also capable of infecting both tumor cells and healthy ones. The presentation of influenza viral antigens in both cases turns the immune system towards the malignant tissue, showing a significant improvement in mice with lung cancer as well as an additive effect with an anti-PD-1 antibody [58].
Despite their therapeutical benefits as vectors, it was observed that, in some situations, previous infections caused by some viruses that express pro-oncogenic proteins contribute to cancer development [59]. These are the cases of Epstein–Barr Virus (EBV), a type-4 herpes virus that has been related to Burkitt lymphoma, or Papillomavirus involvement in Barrett’s dysplasia and esophageal adenocarcinoma [60]. In some cases, during the infection, some parts of the viral genome can be integrated into the genome of the infected cell. This could lead to the expression of viral exogenous proteins that can be presented via MHC-I in the tumor, serving as tumor-specific ones, which would allow targeting malignant cells and developing or repurposing existent vaccines to treat or prevent tumor growth. Recent research [61] showed that an EBV vaccine based on the implementation of four viral glycoprotein-decorating nanoparticles tested in mice could block the viral infection. Additionally, the animals did not develop any lymphomas associated with the virus.
In addition to viruses, another method to address the lack of endogenous antigens in the tumor could be the delivery of MHC-I peptides to tumor cells (Figure 1). During the peptide binding to MHC in the ER, the peptide editor, known as tapasin-related protein (TAPBPR), is essential to shape the final presented peptide [62]. Interestingly, TAPBR has shown the ability to decorate tumor cells with different exogenous peptides present in very low concentrations and to trigger CD8 cytotoxic responses, a promising strategy to make tumors more antigenic [63].
In line with this work, Kavunja and colleagues [64] designed another tool that allowed for the delivery of known MHC-I-dependent antigens to the TME, aiming to elicit an antitumor CD8 response. The authors designed a microparticle capable of delivering peptides in vivo to the tumor and releasing them by degradation in the low pH of the TME. They validated their approach using the ovalbumin-derived peptide SIINFEKL: mice were vaccinated with ovalbumin prior to the inoculation of the tumors, which did not express the protein nor the peptide endogenously. The delivery of the microparticles containing SIINFEKL showed the induction of a potent CD8 immune response which significantly improved the survival of the treated mice [64].
As reviewed in the previous sections, cancer immunotherapy focuses on exploiting the immune system’s endogenous mechanisms to improve its response to malignancies. Once the immune response is unleashed by one of the aforementioned therapeutic interventions, the immune response can expand to other tumor antigens due to a phenomenon known as epitope spreading [65]. This process acts as a cascade expanding the array of antigens that the immune system employs to target the tumor: lysis of cancer cells by CD8+ T lymphocytes through the recognition of an initial peptide permits DCs to obtain access to the dead cells’ cytosolic components. DCs then process and present the new epitopes, which can activate new clones of CD8+ and CD4+ lymphocytes through MHC-I and MHC-II antigenic presentation [66]. The relevance of this phenomenon for cancer was described in mice and some vaccine clinical trials. In animal models, vaccination with ovalbumin (OVA)-expressing cells pulsed with the OVA-derived peptide SIINFEKL induced not only CD8 responses to the initial peptide but to other OVA-derived peptides. Mice thus immunized reject tumors [67], showing the importance of epitope spreading in tumor immunology. Patient data from a phase-I clinical trial in which renal carcinoma patients were vaccinated with DCs loaded with MHC-I and MHC-II-dependent peptides showed a positive response to therapy; of note, the vaccine-induced T-cell responses against other targets that were not included therein. This final observation indicates that epitope spreading may occur, boosting the clinical outcome of DC vaccinations [68].
References
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; Coggeshall, M.; et al. Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544.
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461.
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The Foundations of Immune Checkpoint Blockade and the Ipilimumab Approval Decennial. Nat. Rev. Drug Discov. 2021, 21, 509–528.
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104.
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697.
- Massafra, V.; Tundo, S.; Dietzig, A.; Ducret, A.; Jost, C.; Klein, C.; Kontermann, R.E.; Knoetgen, H.; Steegmaier, M.; Romagnani, A.; et al. Proteolysis-Targeting Chimeras Enhance T Cell Bispecific Antibody-Driven T Cell Activation and Effector Function through Increased MHC Class I Antigen Presentation in Cancer Cells. J. Immunol. 2021, 207, 493–504.
- Del Val, M.; Antón, L.C.; Ramos, M.; Muñoz-Abad, V.; Campos-Sánchez, E. Endogenous TAP-Independent MHC-I Antigen Presentation: Not Just the ER Lumen. Curr. Opin. Immunol. 2020, 64, 9–14.
- Van Hall, T.; Wolpert, E.Z.; Van Veelen, P.; Laban, S.; Van Der Veer, M.; Roseboom, M.; Bres, S.; Grufman, P.; De Ru, A.; Meiring, H.; et al. Selective Cytotoxic T-Lymphocyte Targeting of Tumor Immune Escape Variants. Nat. Med. 2006, 12, 417–424.
- Oliveira, C.C.; Van Hall, T. Importance of TAP-Independent Processing Pathways. Mol. Immunol. 2013, 55, 113–116.
- Garrido, G.; Schrand, B.; Rabasa, A.; Levay, A.; D’Eramo, F.; Berezhnoy, A.; Modi, S.; Gefen, T.; Marijt, K.; Doorduijn, E.; et al. Tumor-Targeted Silencing of the Peptide Transporter Induces Potent Antitumor Immunity. Nat. Commun. 2019, 10, 3773.
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647.
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-Specific MHC-II Expression Represents a Tumour-Autonomous Phenotype and Predicts Response to Anti-PD-1/PD-L1 Therapy. Nat. Commun. 2016, 7, 1–10.
- Andres, F.; Yufeng, L.; Dongquan, C.; William, E.G.; Katherine, L.U.; Natalie, D.M.; Erinn, D.K.; Todd, C.B.; Christos, V.; Donald, J.B.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399.
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; De Lerma Barbaro, A.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R.S. CIITA-Induced MHC Class II Expression in Mammary Adenocarcinoma Leads to a Th1 Polarization of the Tumor Microenvironment, Tumor Rejection, and Specific Antitumor Memory. Clin. Cancer Res. 2006, 12, 3435–3443.
- Nair, S.K.; Boczkowski, D.; Morse, M.; Cumming, R.I.; Lyerly, H.K.; Gilboa, E. Induction of Primary Carcinoembryonic Antigen (CEA)-Specific Cytotoxic T Lymphocytes in Vitro Using Human Dendritic Cells Transfected with RNA. Nat. Biotechnol. 1998, 16, 364–369.
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective Ribosomal Products (DRiPs): A Major Source of Antigenic Peptides for MHC Class I Molecules? J. Immunol. 1996, 157, 1823–1826.
- Anton, L.C.; Yewdell, J.W. Translating : Class Immunosurveillance of Pathogens and Tumors. J. Leukoc. Biol. 2014, 95, 551–562.
- Croft, N.P.; Smith, S.A.; Wong, Y.C.; Tan, C.T.; Dudek, N.L.; Flesch, I.E.A.; Lin, L.C.W.; Tscharke, D.C.; Purcell, A.W. Kinetics of Antigen Expression and Epitope Presentation during Virus Infection. PLoS Pathog. 2013, 9, e1003129.
- Esquivel, F.; Yewdell, J.; Bennink, J. RMA/S Cells Present Endogenously Synthesized Cytosolic Proteins to Class i-Restricted Cytotoxic T Lymphocytes. J. Exp. Med. 1992, 175, 163–168.
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5.
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69.
- Wan, Y.; Wu, C.J. SF3B1 Mutations in Chronic Lymphocytic Leukemia. Blood 2013, 121, 4627–4634.
- Oka, M.; Xu, L.; Suzuki, T.; Yoshikawa, T.; Sakamoto, H.; Uemura, H.; Yoshizawa, A.C.; Suzuki, Y.; Nakatsura, T.; Ishihama, Y.; et al. Aberrant Splicing Isoforms Detected by Full-Length Transcriptome Sequencing as Transcripts of Potential Neoantigens in Non-Small Cell Lung Cancer. Genome Biol. 2021, 22, 1–30.
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic Modulation of RNA Splicing Enhances Anti-Tumor Immunity. Cell 2021, 184, 4032–4047.
- North, K.; Benbarche, S.; Liu, B.; Pangallo, J.; Chen, S.; Stahl, M.; Bewersdorf, J.P.; Stanley, R.F.; Erickson, C.; Cho, H.; et al. Synthetic Introns Enable Splicing Factor Mutation-Dependent Targeting of Cancer Cells. Nat. Biotechnol. 2022, 40, 1103–1113.
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol 2017, 18, 1009–1021.
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from Nonsense-Mediated Decay Associates with Anti-Tumor Immunogenicity. Nat. Commun. 2020, 11, 1–11.
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The Rules and Impact of Nonsense-Mediated MRNA Decay in Human Cancers. Nat. Genet. 2016, 48, 1112–1118.
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of Tumour Immunity by Targeted Inhibition of Nonsense-Mediated MRNA Decay. Nature 2010, 465, 227–230.
- Zhang, Y.; Xie, X.; Yeganeh, P.N.; Lee, D.J.; Valle-Garcia, D.; Meza-Sosa, K.F.; Junqueira, C.; Su, J.; Luo, H.R.; Hide, W.; et al. Immunotherapy for Breast Cancer Using EpCAM Aptamer Tumor-Targeted Gene Knockdown. Proc. Natl. Acad. Sci. USA 2021, 118, e2022830118.
- Meraviglia-Crivelli, D.; Villanueva, H.; Menon, A.P.; Zheleva, A.; Moreno, B.; Villalba-Esparza, M.; Pastor, F. A Pan-Tumor-SiRNA Aptamer Chimera to Block Nonsense-Mediated MRNA Decay (NMD) Inflames and Suppresses Tumor Progression. Mol. Ther. Nucleic Acids 2022.
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting Nonsense-Mediated Decay in Colorectal Cancers with Microsatellite Instability. Oncogenesis 2018, 7, 70.
- Haen, S.P.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards New Horizons: Characterization, Classification and Implications of the Tumour Antigenic Repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610.
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and Putative Neoantigen Load Predict Clinical Benefit of Adoptive T Cell Therapy in Melanoma. Nat. Commun. 2017, 8, 1738.
- Kumari, S.; Sharma, S.; Advani, D.; Khosla, A.; Kumar, P.; Ambasta, R.K. Unboxing the Molecular Modalities of Mutagens in Cancer. Environ. Sci. Pollut. Res. 2021, 1–49.
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413.
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384.
- Almeida, M.V.; Vernaz, G.; Putman, A.L.K.; Miska, E.A. Taming Transposable Elements in Vertebrates: From Epigenetic Silencing to Domestication. Trends Genet. 2022, 38, 529–553.
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973.
- Bhuvanagiri, M.; Lewis, J.; Putzker, K.; Becker, J.P.; Leicht, S.; Krijgsveld, J.; Batra, R.; Turnwald, B.; Jovanovic, B.; Hauer, C.; et al. 5-Azacytidine Inhibits Nonsense-Mediated Decay in a MYC-Dependent Fashion. EMBO Mol. Med. 2014, 6, 1593–1609.
- Ma, R.; Rei, M.; Woodhouse, I.; Ferris, K.; Kirschner, S.; Chandran, A.; Gileadi, U.; Chen, J.-L.; Pereira Pinho, M.; Ariosa-Morejon, Y.; et al. Decitabine Increases Neoantigen and Cancer Testis Antigen Expression to Enhance T-Cell–Mediated Toxicity against Glioblastoma. Neuro. Oncol. 2022, noac107.
- Wong, J.; Gruber, E.; Maher, B.; Waltham, M.; Sabouri-Thompson, Z.; Jong, I.; Luong, Q.; Levy, S.; Kumar, B.; Brasacchio, D.; et al. Integrated Clinical and Genomic Evaluation of Guadecitabine (SGI-110) in Peripheral T-Cell Lymphoma. Leukemia 2022, 36, 1654–1665.
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588.
- Wang, G.; Chow, R.D.; Bai, Z.; Zhu, L.; Errami, Y.; Dai, X.; Dong, M.B.; Ye, L.; Zhang, X.; Renauer, P.A.; et al. Multiplexed Activation of Endogenous Genes by CRISPRa Elicits Potent Antitumor Immunity. Nat. Immunol. 2019, 20, 1494–1505.
- Lhuillier, C.; Formenti, S.C.; Demaria, S.; Lhuillier, C.; Rudqvist, N.; Yamazaki, T.; Zhang, T.; Charpentier, M.; Galluzzi, L.; Dephoure, N.; et al. Radiotherapy-Exposed CD8+ and CD4+ Neoantigens Enhance Tumor Control. J. Clin. Invest. 2021, 131.
- Hietala, A.; Joutsen, J.; Vaarala, S.; Säily, M. A Rare and Complete Response to Combination Therapy with Radiation and Nivolumab in a Patient with Metastatic Urothelial Cancer. BMJ Case Rep. 2022, 15, e246653.
- Giri, A.K.; Aittokallio, T. DNMT Inhibitors Increase Methylation in the Cancer Genome. Front. Pharmacol. 2019, 10, 385.
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767.
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670.
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic Virus Therapy in Cancer: A Current Review. World J. Virol. 2021, 10, 229–255.
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622.
- Lucas, A.; McFadden, G. Secreted Immunomodulatory Viral Proteins as Novel Biotherapeutics. J. Immunol. 2004, 173, 4765–4774.
- Laspidea, V.; Fueyo, J.; Alonso, M.M.; Laspidea, V.; Labiano, S.; Marrodán, L.; Ausejo-mauleon, I.; De Nava, D.; Herrador-cañete, G. Exploiting 4-1BB Immune Checkpoint to Enhance the Efficacy of Oncolytic Virotherapy for Diffuse Intrinsic Pontine Gliomas. JCI Insight 2022, 7, e154812.
- Tian, L.; Liu, T.; Jiang, S.; Cao, Y.; Kang, K.; Su, H.; Ren, G.; Wang, Z.; Xiao, W.; Li, D. Oncolytic Newcastle Disease Virus Expressing the Co-Stimulator OX40L as Immunopotentiator for Colorectal Cancer Therapy. Gene Ther. 2021, 2021, 1–11.
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788.
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral Injection of the Seasonal Flu Shot Converts Immunologically Cold Tumors to Hot and Serves as an Immunotherapy for Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128.
- Tempera, I.; Lieberman, P.M. Oncogenic Viruses as Entropic Drivers of Cancer Evolution. Front. Virol. 2021, 1, 28.
- Cook, K.W.; Durrant, L.G.; Brentville, V.A. Current Strategies to Enhance Anti-Tumour Immunity. Biomedicines 2018, 6, 37.
- Wei, C.J.; Bu, W.; Nguyen, L.A.; Batchelor, J.D.; Kim, J.H.; Pittaluga, S.; Fuller, J.R.; Nguyen, H.; Chou, T.H.; Cohen, J.I.; et al. A Bivalent Epstein-Barr Virus Vaccine Induces Neutralizing Antibodies That Block Infection and Confer Immunity in Humanized Mice. Sci. Transl. Med. 2022, 14, eabf3685.
- Ilca, T.; Boyle, L.H. The Ins and Outs of TAPBPR. Curr. Opin. Immunol. 2020, 64, 146–151.
- Tudor Ilca, F.; Neerincx, A.; Wills, M.R.; De La Roche, M.; Boyle, L.H. Utilizing TAPBPR to Promote Exogenous Peptide Loading onto Cell Surface MHC I Molecules. Proc. Natl. Acad. Sci. USA 2018, 115, E9353–E9361.
- Kavunja, H.W.; Lang, S.; Sungsuwan, S.; Yin, Z.; Huang, X. Delivery of Foreign Cytotoxic T Lymphocyte Epitopes to Tumor Tissues for Effective Antitumor Immunotherapy against Pre-Established Solid Tumors in Mice. Cancer Immunol. Immunother. 2017, 66, 451–460.
- Vanderlugt, C.L.; Miller, S.D. Epitope Spreading in Immune-Mediated Diseases: Implications for Immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95.
- Brossart, P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin. Cancer Res. 2020, 26, 4442–4447.
- El-Shami, K.; Tirosh, B.; Bar-Haîm, E.; Carmon, L.; Vadai, E.; Fridkin, M.; Feldman, M.; Eisenbach, L. MHC Class I-Restricted Epitope Spreading in the Context of Tumor Rejection Following Vaccination with a Single Immunodominant CTL Epitope. Eur. J. Immunol. 1999, 29, 3295–3301.
- Wierecky, J.; Müller, M.R.; Wirths, S.; Halder-Oehler, E.; Dörfel, D.; Schmidt, S.M.; Häntschel, M.; Brugger, W.; Schröder, S.; Horger, M.S.; et al. Immunologic and Clinical Responses after Vaccinations with Peptide-Pulsed Dendritic Cells in Metastatic Renal Cancer Patients. Cancer Res. 2006, 66, 5910–5918.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
705
Revisions:
2 times
(View History)
Update Date:
09 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No