 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rosa M. Ortuño | + 3984 word(s) | 3984 | 2020-10-14 11:48:29 | | | |
| 2 | Rita Xu | + 211 word(s) | 4195 | 2020-10-20 03:38:08 | | |
Video Upload Options
Two series of new hybrid γ/γ-peptides, γ-CC and γ-CT, formed by (1S,2R)-3-amino-2,2-dimethylcyclobutane-1-carboxylic acid joined in alternation to a Nα-functionalized cis- or trans-γ-amino-L-proline derivative, respectively, have been synthesized and evaluated as cell penetrating peptides (CPP) and as selective vectors for anti-Leishmania drug delivery systems (DDS). They lacked cytotoxicity on the tumoral human cell line HeLa with a moderate cell-uptake on these cells. In contrast, both γ-CC and γ-CT tetradecamers were microbicidal on the protozoan parasite Leishmania beyond 25 μM, with significant intracellular accumulation. They were conjugated to fluorescent doxorubicin (Dox) as a standard drug showing toxicity beyond 1 μM, while free Dox was not toxic. Intracellular accumulation was 2.5 higher than with Dox-TAT conjugate (TAT = transactivator of transcription, taken as a standard CPP). The conformational structure of the conjugates was approached both by circular dichroism spectroscopy and molecular dynamics simulations. Altogether, computational calculations predict that the drug-γ-peptide conjugates adopt conformations that bury the Dox moiety into a cavity of the folded peptide, while the positively charged guanidinium groups face the solvent. The favorable charge/hydrophobicity balance in these CPP improves the solubility of Dox in aqueous media, as well as translocation across cell membranes, making them promising candidates for DDS.
1. Introduction
Novel drug delivery systems (DDS) have been developed to achieve a more effective and specific delivery of the drugs to the target organ or cell. One of the major advantages of this approach is to avoid, or at least decrease, the side-effects associated with conventional drugs [1]. In this regard, DDS, together with imaging techniques, are key tools in theranostics or personalized medicine [2][3].
Cell penetrating peptides (CPP) [4][5][6] are potential carriers for DDS. They consist of 5–30 amino acid-long peptides which are capable of transporting the cargo molecule into the intracellular space in the absence of a cognate transporter. At a higher specificity level, the inclusion of an import motif in the CPP may even make it possible to direct the cargo into specific organelles. Either covalent or noncovalent bonding between the cargo molecule and the CPP underlies their use as DDS [7][8][9]. CPP possess appealing features such as good biocompatibility, the potential to fine tune their stability and solubility in biological environments, and potential to create new multifunctional DDS [10]. As such, CPP were implemented as a new tool in the therapeutics of a variety of diseases, with special relevance for cancer [11].
Optimization of the performance of CPP aims to improve both their proteolytic resilience and cell uptake, as well as to achieve minimal toxicity. Endosomal escape ability is also an important characteristic of optimal CPP. Imperviousness to peptidases can be tackled by the inclusion of non-natural or stereochemically modified amino acids, as well as of peptide bond surrogates, in the peptide sequence [12]. Otherwise, this can also be addressed by the introduction of conformational constraints to obtain a more stable secondary structure in the CPP, which is sometimes associated with better cell uptake [12]. For cationic CPP, uptake and toxicity are highly dependent on the number and spatial distribution throughout their sequence. With these two goals in mind, CPP were designed with the inclusion of cyclic amino acids, mainly proline and γ-aminoproline [13][14], helical peptide foldamers [15][16][17], and cyclic peptide backbones [18][19][20].
In recent years, two diastereomeric series of short cell-penetrating hybrid γ/γ-peptides were efficiently prepared through convergent synthesis in solution. These peptides were formed by repetition of a dimeric unit constituted by cis-γ-amino-l-proline, 3, combined with a protected derivative of either (1S,3R)- or (1R,3S)-3-amino-2,2-dimethylcyclobutane-1-carboxylic acid, namely (1S,3R)-γ-CBAA, 1, or (1R,3S)-γ-CBAA, 2, (Figure 1). By high-resolution NMR, these peptides displayed very rigid and compact structures due to the intra- and inter-residue hydrogen-bonded ring formation [21].
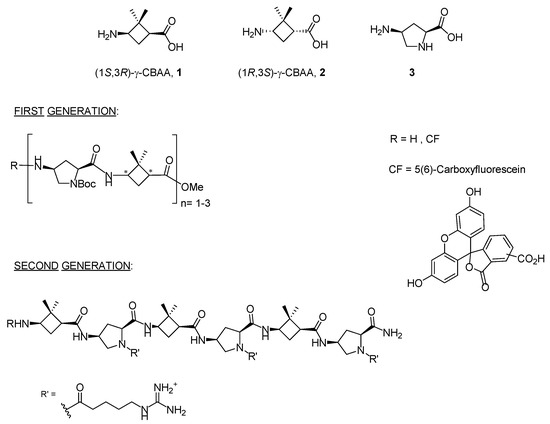
Figure 1. Monomers used in the first and second generations of hybrid cyclobutane/proline γ/γ-peptides labelled with 5(6)-carboxyfluorescein (CF).
Their uptake by HeLa cells increased with the length of the peptide; a five-carbon spacer between the proline Nα atom and the terminal guanidinium group of the side chain was optimal for cell uptake, while the stereochemistry of the γ-CBAA was largely irrelevant. A good polar-hydrophobicity balance was achieved by the alternation of the guanidinium groups and the hydrophobicity of the (gem-dimethyl)cyclobutane ring [21][22]. These cyclobutane-containing CPP showed lower toxicity but similar cell uptake to those made exclusively of γ-amino proline residues [23], likely due to their halved number of guanidinium groups compared with the γ-aminoproline peptides of the same length.
Nowadays, the therapeutic use of CPP-based DDS is mainly focused on cancer, running far beyond examples involving infectious diseases and their causative agents [24]. Nevertheless, infective bacteria, fungi and protozoa are an appealing test for CPP. The cell membrane of these pathogens shows an external anionic hemilayer, in contrast to the zwitterionic one of mammalian cells, privileging interactions of cationic CPP with such pathogens [25]. Furthermore, pathogenic organisms are frequently endowed with a high proteolytic armamentarium as part of their virulence program [26].
A case in point is the human protozoan parasite Leishmania, the causative agent for the wide clinical spectrum of leishmaniasis, a disease with a significant impact on global human health [27]. There is an urgent need for new chemotherapy alternatives for leishmaniasis [28][29][30] to reduce the side effects associated with current chemotherapy, despite the existence of some drugs such as paromomycin [31] or miltefosine [32]. Furthermore, Leishmania is a challenging organism for membrane active peptides [33], including CPP. All the endocytic traffic, a common pathway for CPP uptake, is carried out through the flagellar pocket [34], a specialized region accounting for only 5% of the total surface of the parasite [35]. In addition, the promastigote, responsible for the primary infection in the mammalian host, displays a highly anionic glycocalyx [36], and its main plasma membrane protein is leishmaniolysin, a Zn2+-metalloprotease of broad substrate specificity [37]. The aflagellated amastigote is the pathological form of the parasite in vertebrates, dwelling in the parasitophorus vacuole of the macrophage, a strong proteolytic and nutrient-demanding environment.
There are few examples of the use of CPP in the treatment of Leishmania [38][39][40]. Even so, the use of CPP as DDS on Leishmania parasites has been reported for the delivery of cargoes which are noncovalently complexed with CPP [41][42][43], or as covalent conjugates [44][45][46][47][48].
The anthraquinone doxorubicin (Dox) is broadly used in cancer chemotherapy, and its biological activity is preserved once conjugated to CPP through its amino group [11][49]. As doxorubicin also has leishmanicidal activity [50][51][52] and is a fluorescent compound, it is an excellent molecular beacon to test the functional performance of CPP as functional DDS, but also for an easy track down of intracellular fate and the accumulation of the conjugate.
2. Design and Synthesis of Peptides and Conjugates
Two series of dodecameric and tetradecameric hybrid oligomers (4–11) were synthesized. They were formed by the repetition of a dipeptide motif made up by (1S,3R)-γ-CBAA, 1, [21] linked to a conveniently functionalized cis- or trans-γ-amino-l-proline (γ-CC series and γ-CT series, respectively), (Chart 1). All of them were prepared using standard protocols of solid phase peptide synthesis (SPPS), either in their free N-terminus form or functionalized with a carboxyfluorescein (CF) fluorescent group, as detailed at the Section 3 and the Supplementary Materials (SM).
TAT48–57 (TAT) [53][54][55] was also synthesized as a reference CPP, either with a free or carboxyfluoresceinated N-terminus (see Supplementary Materials).
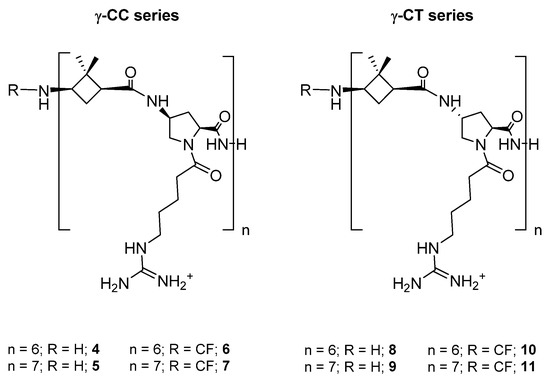
Chart 1. Hybrid peptides of the γ-CC and γ-CT series constituted by repetition of the dipeptide unit γ-CBAA (1)/(cis- or trans)-γ-amino-l-proline, respectively.
Dox was covalently conjugated to γ-CC and γ-CT tetradecameric peptides (Scheme 1), due to their higher internalization rates in Leishmania (Section 2.3). For this purpose (Scheme 1), a cysteine was added to the N-terminal end of the peptides to obtain 12 and 13. Then, they were conjugated to doxorubicin, previously functionalized as 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (MCC), 14, (conjugates 15 and 16). The same conjugation procedure was applied to TAT (see Supplementary Materials for details). To note, a new stereogenic center (marked with an asterisk) was created in the succinimide ring of the linker in the last synthetic step of Dox conjugates, giving two epimers that were used as a mixture. Their influence on the preferred conformations of these conjugates is discussed in Section 2.5 and in the Supplementary Materials.
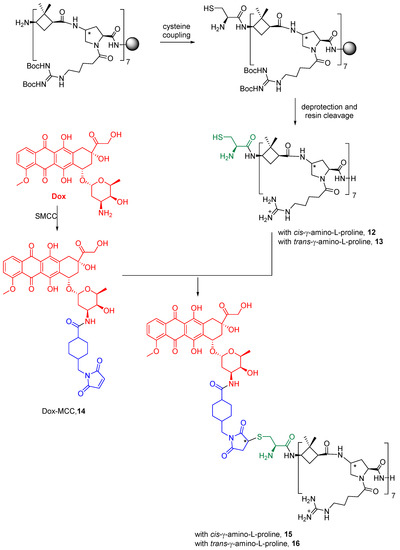
Scheme 1. Conjugation of doxorubicin to γ-CC 15 and γ-CT 16 peptides. The Dox moiety is highlighted in red, the cysteine residue in green, and the linker in blue.
3. Cytotoxicity and Cellular Uptake in HeLa Cells
As a first step to establish the soundness of γ-CC and γ-CT peptides as therapeutic CPP for Leishmania, peptides 4–11 were assayed for their toxicity on HeLa cells, used as a model of mammalian cells. The cellular viability after 24 h incubation with the respective peptide was assessed by the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) [56] (Figure 2).
Even at the highest concentration assayed (50 µM), viability was over 90%, regardless of the peptide stereochemistry (Figure 2). Peptide toxicity was not dependent either on the number of guanidinium groups in the sequence, or on the carboxyfluoresceination of the terminal amino group. In this sense, these results agree with the low toxicity of polyarginine peptides (R)n, up to n ≤ 10 [57].
By flow cytometry, more than 98% of the HeLa cells increased their respective cell-associated fluorescence after incubation with peptides 6, 7, 10 and 11 at the range of concentrations tested (Figure 3). The mean of the population represented in Figure 3 refers either to CF-TAT as a standard CPP, or to CF as an endocytosis marker.
The facts that the performance of TAT on HeLa cells does not increase linearly with peptide concentration (10 μM and higher), and that the cell population becomes heterogeneous with respect to peptide uptake [58], could explain the apparently anomalous results obtained when CF-TAT was used as a reference (Figure 3a). In contrast, the cellular fluorescence for both the γ-CT and γ-CC peptide series increased with concentration and with the length of the peptide when using CF as a control of endocytosis (Figure 3b). Interestingly, in all cases, the fluorescence values for the γ-CT peptides were higher than those of the respective γ-CC homologues.
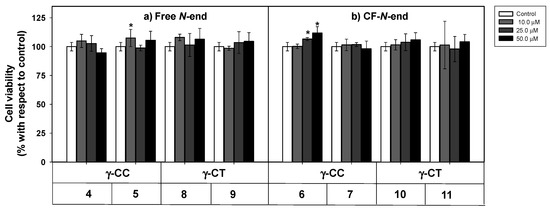
Figure 2. Cytotoxicity of peptides 4–11 on HeLa cells. Peptides with (a) terminal free amino group; (b) terminal amino group conjugated with CF. Cells were incubated with the respective peptide for 24 h and their viability assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. Cell viability was expressed as the percentage of MTT reduction with respect to control cells ± SD. Statistical significance with respect to control cells: p ≤ 0.05 (*). Samples were made in quadruplicate, and experiments were repeated thrice independently.
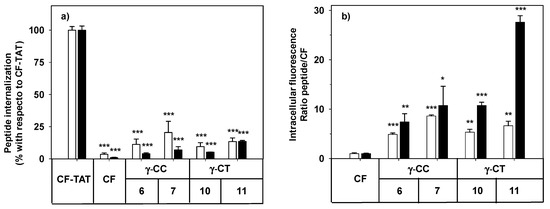
Figure 3. Cellular internalization of carboxyfluoresceinated peptides 6, 7, 10, and 11 normalized (a) with respect to CF-TAT (100%), and (b) as a fluorescence ratio of the peptides with respect to CF as a control of endocytosis (CF fluorescence = 1 a.u.). HeLa cells were incubated with the respective peptide at 10 (empty column), or 25 µM (black column) for 24 h, and the level of fluorescence associated with the cells was assessed by flow cytometry (λEXC = 488 nm and λEM = 530 nm). Results were expressed as mean ± SD. Samples were made in triplicate (p ≤ 0.05, (*); p ≤ 0.01, (**); p ≤ 0.001, (***)). Three independent experiments were carried out.
The cell internalization of peptides 7 and 11 in HeLa cells was evidenced by confocal microscopy under identical conditions to flow cytometry. The intracellular fluorescence showed a spotted cytoplasmic pattern after incubation with the peptides at 25 μM peptide (Figure 4). Confocal 3D ruled out their mere association with the plasma membrane.
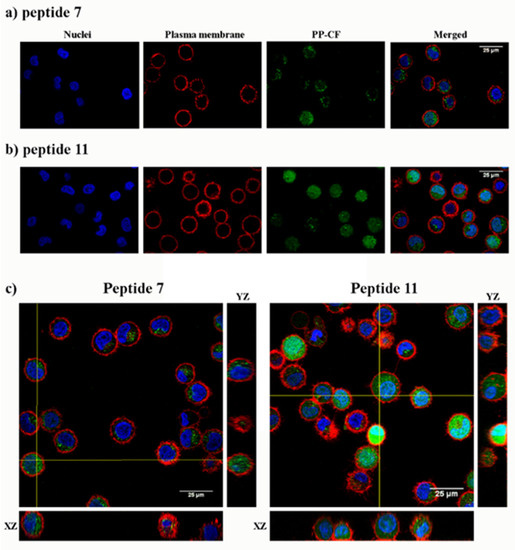
Figure 4. Confocal microscopy of HeLa cells incubated with the carboxyfluoresceinated γ-CC 7 and γ-CT 11 tetradecameric peptides. (a) Peptide γ-CC 7. (b) Peptide γ-CT 11. (c) Orthogonal projections highlight the internalization (XZ and YZ) of both peptides. HeLa cells were incubated with the respective CF peptides (2 h, 37 °C; 25 µM), (green fluorescence λEXC = 488 nm; λEM = 510 nm), and additionally stained with CellMask, deep red (red fluorescence, λEXC = 658 nm; λEM = 690 nm) and Hoechst (blue fluorescence, λEXC = 405 nm; λEM = 460 nm) as a plasma membrane and nucleic acids markers, respectively. Magnification bar: 25 µm.
4. Uptake, Microbicidal Activity and Intracellular Location of Peptides on Leishmania Parasites
Once the lack of toxicity of γ-CPP and their low uptake in mammalian cells compared to TAT had been demonstrated, their performance in Leishmania was examined as a model of microorganisms with anionic plasma and endowed with a high proteolytic level. To this end, peptides were assayed for viability, uptake and location on the protozoan Leishmania parasites.
The cellular viability in the presence of the peptides was assessed by the MTT reduction at both stages of the parasite after 4 h incubation (Figure 5).
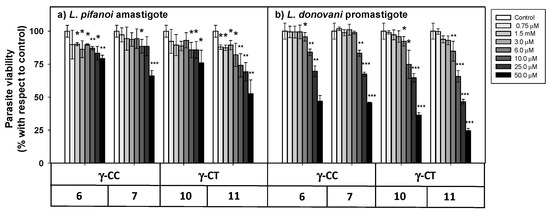
Figure 5. Viability of Leishmania sp. parasites after 4 h incubation with peptides 6, 7, 10 and 11. (a) L. pifanoi axenic amastigotes. (b) L. donovani promastigotes. Axenic Leishmania parasites (20 × 106 cells/mL) were incubated with the peptides, and MTT reduction measured immediately after 4 h incubation. Viability was represented as the percentage of MTT reduction (mean ± SD). Statistical significance with respect to control parasites: p ≤ 0.05, (*); p ≤ 0.01, (**); p ≤ 0.001, (***). Results are representative of one out of three independent experiments. Samples were made in triplicate.
The loss of parasite viability due to peptides was concentration-dependent, with promastigotes (Figure 5b) being more susceptible than amastigotes (Figure 5a). The γ-CT series showed higher toxicity than its respective γ-CC counterparts. This trend was especially noticeable for peptide concentrations over 25 μM. Concerning toxicity, the tetradecapeptides (7 and 11) were slightly more toxic than the corresponding dodecamers (6 and 10), likely due to the increase of their cationic character [59]. Thus, these γ-CPP behave as mild leishmanicidal agents.
Next, the internalization of the carboxyfluoresceinated CPP into promastigotes and amastigotes was measured by flow cytometry, and their values compared to that of CF-TAT (100%). As shown in Figure 6, a similar trend was observed for both parasite systems. The fluorescence values of dodecamers 6 and 10 were relatively similar or up to 30% lower than that of CF-TAT, while parasites incubated with tetradecamers 7 and 11 showed higher fluorescence than CF-TAT.
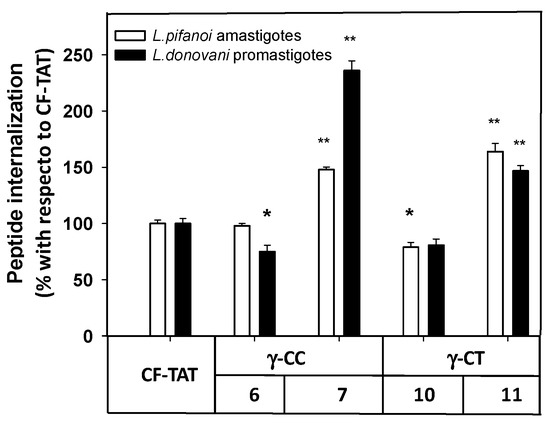
Figure 6. Flow cytometry of carboxyfluoresceinated 6, 7, 10 and 11 and TAT48–57 peptides associated with Leishmania pifanoi amastigotes and Leishmania donovani promastigotes. Parasites (20 × 106 cells/mL) were incubated for 4 h with the respective CF-peptides at 10 μM. Afterwards, peptide association to parasites was assessed by flow cytometry (λEXC = 488 nm, λEM = 519 nm) and represented as the percentage with respect to TAT-CF ± SD. White bars: Leishmania pifanoi axenic amastigotes; black bar: Leishmania donovani promastigotes. Statistical significance was referred with respect to the uptake of carboxyfluoresceinated TAT: p ≤ 0.05, (*); p ≤ 0.01, (**). Results are representative of one out of three independent experiments. Samples were made in triplicate.
Under the same conditions, the intensity of the fluorescence was much higher in promastigotes than in amastigotes (data not shown), likely due to their much smaller cellular size; consequently, the promastigotes were chosen for the studies with confocal microscopy.
Thus, these γ-CPP are potential cargo carriers at concentrations ≤25 μM; over this value, they behave as leishmanicidal agents on their own. This, together with their nil toxicity and poor uptake in mammalian cells, makes them amenable as DDS systems.
The internalization of these peptides in Leishmania donovani promastigotes was assessed by confocal microscopy on those peptides with the best results in flow cytometry. Thus, promastigotes were incubated with peptides 6, 7, 10 and 11 as well as TAT (Figure 7) at a final concentration of 10 μM for 2 h at 26 °C. DAPI was used as a nucleic acid dye to stain the nucleus and the kinetoplast (blue fluorescence) (Figure 7).
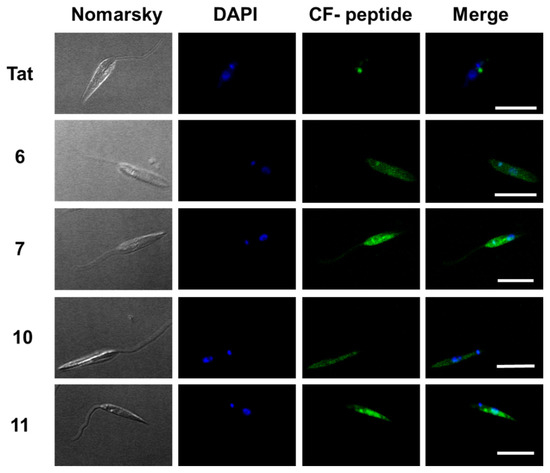
Figure 7. Confocal microscopy of Leishmania donovani promastigotes incubated with the selected γ/γ-peptides and TAT. Parasites were incubated with the corresponding peptides (10 μM final concentration, 2 h, 26 °C). Afterwards, they were observed without further fixation. CF-peptides (green fluorescence: λEXC = 488 nm/λEM = 519 nm). DAPI (DNA probe, blue fluorescence: λEXC = 358 nm/λEM = 461 nm). Magnification bar = 10 μm.
According to Figure 7, the peptides accumulated inside the promastigote. The pattern was not homogenous. Some dotted areas showed higher fluorescence intensity, with a special relevance near the flagellar pocket, the specialized area of Leishmania in charge of all endocytic and exocytic traffic, indicating endocytic CPP uptake. Under these conditions, the nucleus excluded peptides.
In contrast to HeLa uptake, γ-CPP exceeded TAT uptake on Leishmania. In other words, an unexpected selectivity towards the parasite was found for these peptides, which supports their potential as selective vectors in DSS for this protozoan in its vertebrate host. A higher selectivity threshold for DSS against the parasite is of special relevance, due to the feasible avoidance of side-effects associated with the current leishmanicidal drugs in Leishmania [60], and the probable lowering of the selectivity index for the payload with respect to its administration as free drug.
Due to their higher accumulation both in Leishmania and HeLa cells, tetradecamers 7 and 11 were selected as vehicles for Dox-conjugation (conjugates 15 and 16, respectively), in order to be applied as DDS on Leishmania. Figure 8 shows the viability of the parasites when treated with these conjugates.
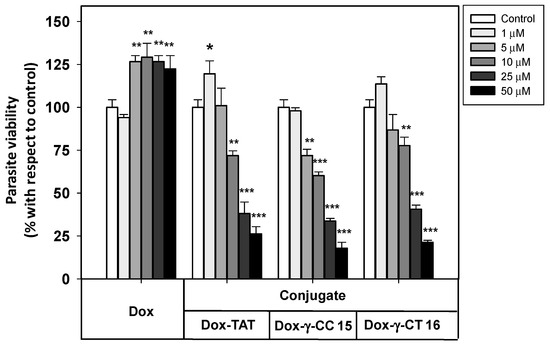
Figure 8. Viability of Leishmania donovani promastigotes treated with Dox-conjugates 15 and 16 along with Dox and Dox-TAT. Parasites were incubated with the respective reagents (20 × 106 cells/mL, 4 h, 26 °C), and MTT reduction measured afterwards. Parasite viability was represented as the percentage of MTT reduction ± SD with respect to untreated parasites. Statistical significance with respect to control parasites: p ≤ 0.05 (*); p ≤ 0.01 (**); p ≤ 0.001 (***). Samples were made in triplicate. Results from one out of three independent experiments were represented.
Free doxorubicin did not show any toxicity towards Leishmania donovani promastigotes at the full range of concentrations assayed, which avoided the leishmanicidal effect caused by free doxorubicin at longer incubation times (<12 h).
In contrast, the Dox-conjugates 15 and 16 showed an increasing toxicity at concentrations ≥ 1 μM. The peptide γ-CC 15 was shown to be slightly more toxic than γ-CT 16 at 5, 10 and 25 μM. MD simulations could account for such a difference (see below). In addition, Dox-TAT toxicity was like that of 15 and 16.
The uptake of free doxorubicin and of their peptide conjugates in L. donovani promastigotes was quantified by flow cytometry at 5 and 10 μM, measured at 2 and 4 h incubations (Figure 9).
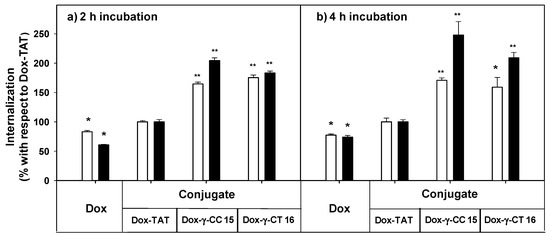
Figure 9. Uptake of free- and peptide-conjugated doxorubicin by Leishmania donovani promastigotes at (a) 2 h incubation, (b) 4 h incubation. Doxorubicin uptake was quantified by flow cytometry using the doxorubicin fluorescence setting (λEXC = 488 nm; λEM = 515 nm) after incubation of the parasites (20 × 106 cells/mL, 26 °C in HBSS-Glc) for (a) 2 h, and (b) 4 h. Data were represented as percentage ± SD with respect to the uptake of the Dox-TAT conjugate. Reagent concentration: 5 μM (white columns); 10 µM (black columns). Samples were made by duplicate (p ≤ 0.05, (*); p ≤ 0.01, (**)). Dox: doxorubicin; HBSS-Glc: Hanks buffered saline solution and glucose.
Dox-conjugates 15 and 16 showed better internalization than Dox-TAT, a trend that was observed with parent peptides 7 and 11 (see Figure 6) despite the modifications introduced (doxorubicin conjugation plus the additional cysteine). Longer incubation did not substantially improve the uptake for 15 and 16 at 5 μM, but it did at 10 μM. Under these conditions, the accumulation of 15 was 2.5-fold higher than that of Dox-TAT.
5. CD Spectroscopy
The CD of γ-CC 5 and γ-CT 9 conjugates showed respective monosignated spectra with λmax at 215 and 212 nm (see Figure S1 in Supplementary Materials); nevertheless, an accurate insight into their folding from these data was insufficient, even when some defined conformational bias for oligomers consisting of γ-amino acids was published [61]. To make these aspects clear, molecular modeling studies were undertaken.
6. Molecular Modeling
The hydrophobicity of amphipathic CPP [62][63] is crucial for their high affinity binding to lipid membranes, including those from internalized vesicles [64][65]. In addition, the creation of high-density positive areas in amphipatic cationic peptides favors their specific binding into anionic biopolymers, such as the cell membranes of pathogens [66].
Therefore, molecular dynamics (MD) simulations were carried out on γ-CC 5 and γ-CT 9 peptides under an explicit solvent scenario. The goals were to shed light on the folding of peptides in aqueous solution and to determine how the arrangement of positive charges could be involved in cellular binding. In addition, the role of their respective cargo molecules (doxorubicin and carboxyfluorescein) in the conformation of the conjugates was also examined. For doxorubicin conjugates, both (R) and (S) epimers were considered (see Supplementary Materials for details).
The trajectories of both CC and CT diastereomeric peptides converged into defined folded states after ~40 ns. In all of them, a hydrophobic core was formed and the polar guanidinium groups of the positively charged residues faced the solvent. A double hairpin motif was predominant for γ-CC 5 (Figure 10a), promoted mostly by diverse inter-residue hydrogen-bonds (Figure S7a). These hairpins, and also β-sheet-like structures, have been observed for similar peptides based on cis-γ-amino-L-proline [67]. For the conjugate (R)-Dox-γ-CC, (R)-15, a stable conformation occurred after ~75 ns, with the Dox moiety being partially buried in the main peptide scaffold (Figure 10b). This perturbation of the original double hairpin by the presence of Dox is the consequence of long-range hydrogen-bonding between the primary hydroxyl group of the cyclohexane side-chain in the Dox moiety and Nα-side-chain CO in proline residue i = 8 (Figure S7b).
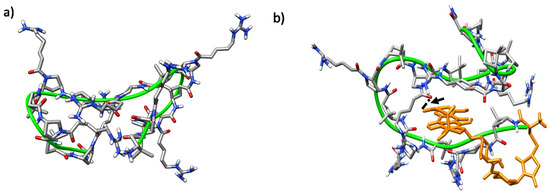
Figure 10. Representative MD conformations for (a) peptide γ-CC 5 and (b) conjugate (R)-Dox-γ-CC, (R)-15. The peptide scaffold is highlighted by the green ribbon; Dox is represented in orange; the arrow points out long-range hydrogen-bonding between the primary hydroxyl group of the cyclohexane side-chain in Dox moiety and the Nα-side chain CO in proline residue i = 8.
A related conformation was the most stable for epimer (S)-Dox-γ-CC, (S)-15; the Dox moiety was folded towards the peptide backbone cavity with the establishment of a long-range hydrogen bond between the hydroxyl group of the amino sugar ring of the Dox-moiety and proline residue i = 12 (Figures S6 and S7c). The hairpin-like conformation was also obtained, along with a laminar one, when the conjugate CF-γ-CC 7 was considered (Figures S8 and S9).
On the other hand, peptide γ-CT 9 adopted a stable right-handed helix structure that was massively occupied (77%) along the trajectory (Figure 11a herein and Figure S10). This folding was driven by intra-residue hydrogen bonds formed by the NH and the carbonyl group in the γ-CBAA units, and inter-residue ones formed between the protons of the guanidinium group in an i residue and the proline Nα-side-chain C=O at I + 4 (Figure 11a).
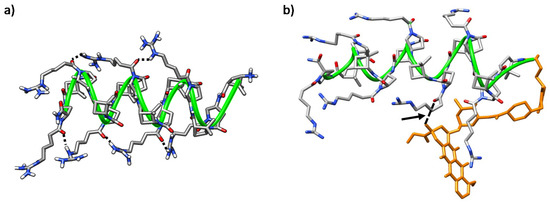
Figure 11. Representative MD conformations for (a) peptide γ-CT 9, inter-residue interactions are marked; and (b) conjugate (R)-Dox-γ-CT, (R)-16. The peptide scaffold is highlighted by the green ribbon; Dox is represented in orange; the arrow points out long-range hydrogen-bonding between the tertiary hydroxyl group of the cyclohexane ring in the Dox-moiety and the Nα-side chain CO in proline residue i = 6.
Helical conformations were also predominant in both (R) and (S) isomers of conjugate Dox-γ-CT 16; in both cases, the Dox moiety faced the peptide backbone, becoming partially hidden. Nevertheless, this interaction is dependent on the configuration at the epimeric center of the linker. In the (R) epimer, a relevant long-range hydrogen bond between the hydroxyl group of the Dox cyclohexane moiety with the Nα-side-chain C=O in proline residue i = 6 was established (Figure 11b and Figure S11b). In contrast, this hydrogen bond was not relevant for the (S) epimer (Figure S11a,c), and doxorubicin was buried in the core of the peptide, probably to minimize its hydrophobic interactions with the aqueous medium. This feature could account for the slight difference of toxicity observed for both conjugates Dox-γ-CC 15 and Dox-γ-CT 16, as noted in Section 2.3.
Moreover, the stability of the helical structure of CF-γ-CT 11 was maintained along 450 ns in the simulation, despite its CF terminal moiety, located at the periphery of the peptide backbone (Figure S12).
Therefore, MD highlights the critical role of the cis/trans stereochemistry of the γ-amino-l-proline monomer for the folding of γ-CC and γ-CT peptide series. Although a specific conformation is not unequivocally associated with a better uptake, good cellular uptake has been reported for helical, hairpin and sheet-like peptides [34][68]. In addition, it is well documented that the delocalized charge of the guanidinium group endows a high membrane-translocation efficiency upon a variety of arginine-rich CPP [68][69][70]. In our case, the conjugation of doxorubicin to the N-terminus of the peptide induced the hydrophobic collapse of the skeleton around the drug, which become buried, regardless of the diastereomeric series or the configuration of epimeric center. As this folding exposes the guanidinium group of the side-chains to the external aqueous medium, better membrane translocation is assumed.
7. Highlights and Conclusions
Cell-penetrating peptides resilient to proteolytic degradation are especially useful for their application into proteolytic environments. Two diastereomeric series of new dodecameric and tetradecameric hybrid γ/γ-peptides, γ-CC and γ-CT, formed by repetition of a dipeptide unit constituted by a chiral cyclobutane amino acid, and either a Nα-functionalized cis- or trans-γ-amino-L-proline derivative, respectively, were synthesized and evaluated as CPP on mammalian cells and Leishmania, an obligate intracellular parasite of the macrophages. The γ/γ-peptides supersede TAT respect to their penetration into Leishmania parasites, while maintaining a slow toxicity profile on mammalian cells. In addition, these peptides behave as mild leishmanicidal agents at higher concentrations. This specific toxicity of the γ/γ-peptides towards Leishmania is a “must” for their use as CPP against the parasite.
These peptides were used to ferry doxorubicin into the tetradecameric Dox-γ-CC conjugate delivered, showing a significant and faster leishmanicidal blow than the free drug. The molecular modeling stresses that the adoption of a well-defined conformation for the tetradecamers is highly influenced by the choice of the diastereomeric unit, even when the impact of the conformation on the leishmanicidal activity is not dramatic. An important fact from MD is that the doxorubicin moiety becomes wrapped by the peptide chain, regardless of the diastereomerism of the peptides employed, hidden from the aqueous environment while exposing the polar guanidinium groups at the periphery of the peptide, and improving the solubility of the drug.
References
- Hossen, S.; Hossain, M.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18.
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The smart drug delivery system and its clinical potential. Theranostics 2016, 6, 1306–1323.
- Schmitt, J.; Heitz, V.; Sour, A.; Bolze, F.; Kessler, P.; Flamigni, L.; Ventura, B.; Bonnet, C.S.; Toth, E. A Theranostic agent combining a two-photon-absorbing photosensitizer for photodynamic therapy and a gadolinium(III) complex for MRI detection. Chem. Eur. J. 2016, 22, 2775–2786.
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating Peptides. Trends Pharmacol. Sci. 2000, 21, 99–103.
- Langel, Ü. Cell-Penetrating Peptides in Processes and Applications; CRC Press Pharmacology and Toxicology Series: Boca Raton, FL, USA, 2002.
- Lundberg, P.; Langel, Ü. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233.
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138.
- Koren, E.; Torchillin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393.
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994.
- Zhang, D.; Wang, J.; Xu, D. Cell penetrating peptides as noninvasive transmembrane vectors for the development of new functional drug delivery systems. J. Control. Release 2016, 229, 130–139.
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76.
- Fominaya, J.; Bravo, J.; Rebollo, A. Strategies to stabilize cell penetrating peptides for in vivo applications. Ther. Del. 2015, 6, 1171–1194.
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484.
- Dobitz, S.; Aronoff, M.R.; Wennemers, H. Oligoprolines as molecular entities for controlling distance in biological and material sciences. Acc. Chem. Res. 2017, 50, 2420–2428.
- Potocky, T.B.; Menon, A.K.; Gellman, S.H. Effects of conformational stability and geometry of guanidinium display on cell entry by β-peptides. J. Am. Chem. Soc. 2005, 127, 3686–3687.
- Nagel, Y.A.; Raschle, P.S.; Wennemers, H. Effect of preorganized charge-display on the cell-penetrating properties of cationic peptides. Angew. Chem. 2017, 56, 122–126.
- Tian, Y.; Zeng, X.; Li, J.; Jiang, Y.; Zhao, H.; Wang, D.; Huang, X.; Li, Z. Achieving enhanced cell penetration of short conformationally constrained peptides through amphiphilicity tuning. Chem. Sci. 2017, 8, 7576–7581.
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453.
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. 2015, 54, 1950–1953.
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612.
- Gutiérrez-Abad, R.; Carbajo, D.; Nolis, P.; Acosta-Silva, C.; Cobos, J.A.; Illa, O.; Royo, M.; Ortuño, R.M. Synthesis and structural study of highly constrained hybrid cyclobutane-proline γ,γ-peptides. Amino Acids 2011, 41, 673–686.
- Gorrea, E.; Carbajo, D.; Gutiérrez-Abad, R.; Illa, O.; Branchadell, V.; Royo, M.; Ortuño, R.M. Searching for new cell-penetrating agents: Hybrid cyclobutane–proline γ,γ-peptides. Org. Biomol. Chem. 2012, 10, 4050–4057.
- Farrera-Sinfreu, J.; Giralt, E.; Castel, S.; Albericio, F.; Royo, M. Cell-penetrating cis-γ-amino-L-proline-derived peptides. J. Am. Chem. Soc. 2005, 127, 9459–9468.
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429.
- Rivas, L.; Nácher-Vázquez, M.; Andreu, D. The Physical Matrix of the Plasma Membrane as a Target: The Charm of Drugs with Low Specificity. In Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; The Royal Society of Chemistry: London, UK, 2018; pp. 248–281.
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331.
- World Health Organization. Available online: http://www.who.int/leishmaniasis/en/ (accessed on 7 September 2020).
- Chakravarty, J.; Sundar, S. Drug resistance in leishmaniasis. J. Glob. Infect. Dis. 2010, 2, 167–176.
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human african trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347.
- Hefnawy, A.; Berg, M.; Dujardin, J.-C.; De Muylder, G. Exploiting knowledge on Leishmania drug resistance to support the quest for new drugs. Trends Parasitol. 2017, 33, 162–174.
- Paromomycin SULFATE. Available online: https://www.webmd.com/drugs/2/drug-5160/paromomycin-oral/details#side-effects (accessed on 4 September 2020).
- Drugs and Supplements Miltefosine (Oral Route). Available online: htpps://www.mayoclinic.org/drugs-supplements/miltefosine-oral-route/side-effects/drg-20095231 (accessed on 4 September 2020).
- Naderer, T.; Vince, J.E.; McConville, M.J. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr. Mol. Med. 2004, 4, 649–665.
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane crossing and membranotropic activity of cell-penetrating peptides: Dangerous liaisons? Acc. Chem. Res. 2017, 50, 2968–2975.
- Morgan, G.W.; Hall, B.S.; Denny, P.W.; Carrington, M.; Field, M.C. The Kinetoplastida endocytic apparatus. Part I: A dynamic system for nutrition and evasion of host defences. Trends Parasitol. 2002, 18, 491–496.
- Forestier, C.L.; Gao, Q.; Boons, G.J. Leishmania lipophosphoglycan: How to establish structure-activity relationships for this highly complex and multifunctional glycoconjugate? Front. Cell. Infect. Microbiol. 2015, 4, 193.
- Olivier, M.; Atayde, V.D.; Isnard, A.; Hassani, K.; Shio, M.T. Leishmania virulence factors: Focus on the metalloprotease GP63. Microbes Infect. 2012, 14, 1377–1389.
- Luque-Ortega, J.R.; Rivas, L. Characterization of the Leishmanicidal Activity of Antimicrobial Peptides. In Antimicrobial Peptides. Methods in Molecular Biology (Methods and Protocols); Giuliani, A., Rinaldi, A., Eds.; Humana Press: Totowa, NJ, USA, 2010; Volume 618, 393–420
- Silva, T.; Abengózar, M.A.; Fernández-Reyes, M.; Andreu, D.; Nazmi, K.; Bolscher, J.G.M.; Bastos, M.; Rivas, L. Enhanced leishmanicidal activity of cryptopeptide chimeras from the active N1 domain of bovine lactoferrin. Amino Acids 2012, 43, 2265–2277.
- Ruiz-Santaquiteria, M.; Sánchez-Murcia, P.A.; Toro, M.A.; de Lucio, H.; Gutiérrez, K.J.; de Castro, S.; Carneiro, F.A.C.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; et al. First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem. 2017, 135, 49–59.
- Keller, A.-A.; Mussbach, F.; Breitling, R.; Hemmerich, P.; Schaefer, B.; Lorkowski, S.; Reissmann, S. Relationships between cargo, cell penetrating peptides and cell type for uptake of non-covalent complexes into live cells. Pharmaceuticals 2013, 6, 184–203.
- Keller, A.A.; Breitling, R.; Hemmerich, P.; Kappe, K.; Braun, M.; Wittig, B.; Schaefer, B.; Lorkowski, S.; Reissmann, S. Transduction of proteins into Leishmania tarentolae by formation of non‐covalent complexes with cell‐penetrating peptides. J. Cell. Biochem. 2014, 115, 243–252.
- Keller, A.A.; Scheiding, B.; Breitling, R.; Licht, A.; Hemmerich, P.; Lorkowski, S.; Reissmann, S. Transduction and transfection of difficult‐to‐transfect cells: Systematic attempts for the transfection of protozoa Leishmania. J. Cell. Biochem. 2019, 120, 14–27.
- Kóczán, G.; Ghose, A.C.; Mookerjee, A.; Hudecz, F. Methotrexate conjugate with branched polypeptide influences Leishmania donovani infection in vitro and in experimental animals. Bioconjug. Chem. 2002, 13, 518–524.
- Luque-Ortega, J.R.; de la Torre, B.G.; Hornillos, V.; Bart, J.-M.; Rueda, C.; Navarro, M.; Amat-Guerri, F.; Ulises Acuña, A.; Andreu, D.; Rivas, L. Defeating Leishmania resistance to miltefosine (hexadecylphosphocholine) by peptide-mediated drug smuggling: A proof of mechanism for trypanosomatid chemotherapy. J. Control. Release 2012, 161, 835–842.
- Torre, B.G.; Hornillos, V.; Luque-Ortega, J.R.; Abengozar, M.A.; Amat-Guerri, F.; Ulises Acuna, A.; Rivas, L.; Andreu, D. A BODIPY-embedding miltefosine analog linked to cell-penetrating TAT(48-60) peptide favors intracellular delivery and visualization of the antiparasitic drug. Amino Acids 2014, 46, 1047–1058.
- de Lucio, H.; Gamo, A.M.; Ruiz-Santaquiteria, M.; de Castro, S.; Sánchez-Murcia, P.A.; Toro, M.A.; Gutiérrez, K.J.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; et al. Improved proteolytic stability and potent activity against Leishmania infantum trypanothione reductase of α/β-peptide foldamers conjugated to cell-penetrating peptides. Eur. J. Med. Chem. 2017, 140, 615–623.
- Defaus, S.; Gallo, M.; Abengózar, M.A.; Rivas, L.; Andreu, D. A synthetic strategy for conjugation of paromomycin to cell-penetrating TAT(48-60) for delivery and visualization into Leishmania parasites. Int. J. Pept. 2017, 2017, doi:10.1155/2017/4213037.
- Hua, Q.; Quiang, Z.; Chu, M.; Shi, D.; Ren, J. Polymeric drug delivery system with actively targeted cell penetration and nuclear targeting for cancer therapy. ACS Appl. Bio Mater, 2019, 2, 1724–1731.
- Sett, R.; Basu, N.; Ghosh, A.K.; Das, P.K. Potential of doxorubicin as an antileishmanial agent. J. Parasitol. 1992, 78, 350–354. .
- Mukherjee, S.; Das, L.; Kole, L.; Karmakar, S.; Datta, N.; Das, P.K. Targeting of parasite-specific immunoliposome-encapsulated doxorubicin in the treatment of experimental visceral leishmaniasis. J. Infect. Dis. 2004, 189, 1024–1034.
- Sundar, S.; Chakravarty, J. Investigational drugs for visceral leishmaniosis. Expert Opin. Investig. Drugs 2015, 24, 43–59.
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus TAT trans-activator protein. Cell 1988, 55, 1179–1188.
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the TAT protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193.
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 TAT protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017.
- Mosnann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays J. Immunol. Methods 1983, 65, 55–63.
- Futaki, S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug. Deliv. Rev. 2005, 57, 547–558.
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866.
- Aguilera, T.A.; Timmers, M.M.; Olson, E.S.; Jiang, T.; Tsien, R.Y. Systemic in vivo distribution of activatable cell penetrating peptides is superior to cell penetrating peptides. Integr. Biol. 2009, 1, 371–381.
- WHO Technical report series. In Proceedings of the Control of the leishmaniases: Report of a Meeting of the WHO Expert Commitee on the Control of Leishmaniases, Geneva, Switzerland, 22–26 March 2010.
- Seebach, D.; Brenner, M.; Rueping, M.; Jaun, B. γ2-, γ 3-, and γ 2,3,4-Amino acids, coupling to -hexapeptides: CD spectra, NMR solution and X-ray Crystal structures of γ -peptides. Chem. Eur. J. 2002, 8, 573–584.
- Deshayes, S.; Plénat, T.; Aldrian-Herrada, G.; Divita, G.; Le Grimellec, C.; Heitz, F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry 2004, 43, 7698–7706.
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597.
- Zaro, J.L.; Vekich, J.E.; Tran, T.; Shen, W.-C. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol. Pharm. 2009, 6, 337–344.
- Jiao, C.Y.; Delaroche, D.; Durlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 2009, 284, 33957–33965.
- Pae, J.; Liivamägi, L.; Lubenets, D.; Arukuusk, P.; Langel, Ü.; Pooga, M. Glycosaminoglycans are required for translocation of amphipathic cell-penetrating peptides across membranes. Biochim. Biophys. Acta 2016, 1858, 1860–1867
- Farrera-Sinfreu, J.; Zaccaro, L.; Vidal, D.; Salvatella, X.; Giralt, E.; Pons, M.; Albericio, F.; Royo, M. A new class of foldamers based on cis-γ-amino-L-proline. J. Am. Chem. Soc. 2004, 126, 6048–6057.
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607.
- Izawa, H.; Kinai, M.; Ifuku, S.; Morimoto, M.; Saimoto, H. Guanidinylated chitosan inspired by arginine-rich cell-penetrating peptides. Int. J. Biol. Macromol. 2019, 125, 901–905.
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P., Arginine "magic": Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc. Chem. Res. 2018, 51, 1455–1464.

