Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pawel Kozyra | -- | 4570 | 2022-08-31 09:38:54 | | | |
| 2 | Conner Chen | -27 word(s) | 4543 | 2022-09-01 10:16:44 | | | | |
| 3 | Conner Chen | + 4 word(s) | 4547 | 2022-09-01 12:03:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kozyra, P.; Pitucha, M. Novel Agent with the Terminal Phenoxy Group. Encyclopedia. Available online: https://encyclopedia.pub/entry/26715 (accessed on 26 July 2026).
Kozyra P, Pitucha M. Novel Agent with the Terminal Phenoxy Group. Encyclopedia. Available at: https://encyclopedia.pub/entry/26715. Accessed July 26, 2026.
Kozyra, Paweł, Monika Pitucha. "Novel Agent with the Terminal Phenoxy Group" Encyclopedia, https://encyclopedia.pub/entry/26715 (accessed July 26, 2026).
Kozyra, P., & Pitucha, M. (2022, August 31). Novel Agent with the Terminal Phenoxy Group. In Encyclopedia. https://encyclopedia.pub/entry/26715
Kozyra, Paweł and Monika Pitucha. "Novel Agent with the Terminal Phenoxy Group." Encyclopedia. Web. 31 August, 2022.
Copy Citation
The terminal phenoxy group is a moiety of many drugs in use today. Numerous literature reports indicated its crucial importance for biological activity; thus, it is a privileged scaffold in medicinal chemistry. Most often, the presence of the phenoxy moiety provided the chances for the compound to match the target, ensuring selectivity, the π–π interaction, or increase the ability to form the hydrogen bonds by the oxygen ether atom.
phenoxy group
drug scaffold
anticancer activity
neurological disorder
anti-HIV activity
antimicrobial
analgesic activity
lymphoma
antidiabetic
adrenergic receptor activity
1. Neurological Disorders
In 2016, neurological disorders were the second most common cause of death [1]. On the one hand, people have depressive disorders, anxiety disorders, and phobias, and on the other, neurodegenerative diseases. The most major are Alzheimer’s and Parkinson’s [2]. The novel potential agents for neurological disorder bearing a phenoxy group are presented in Figure 1.
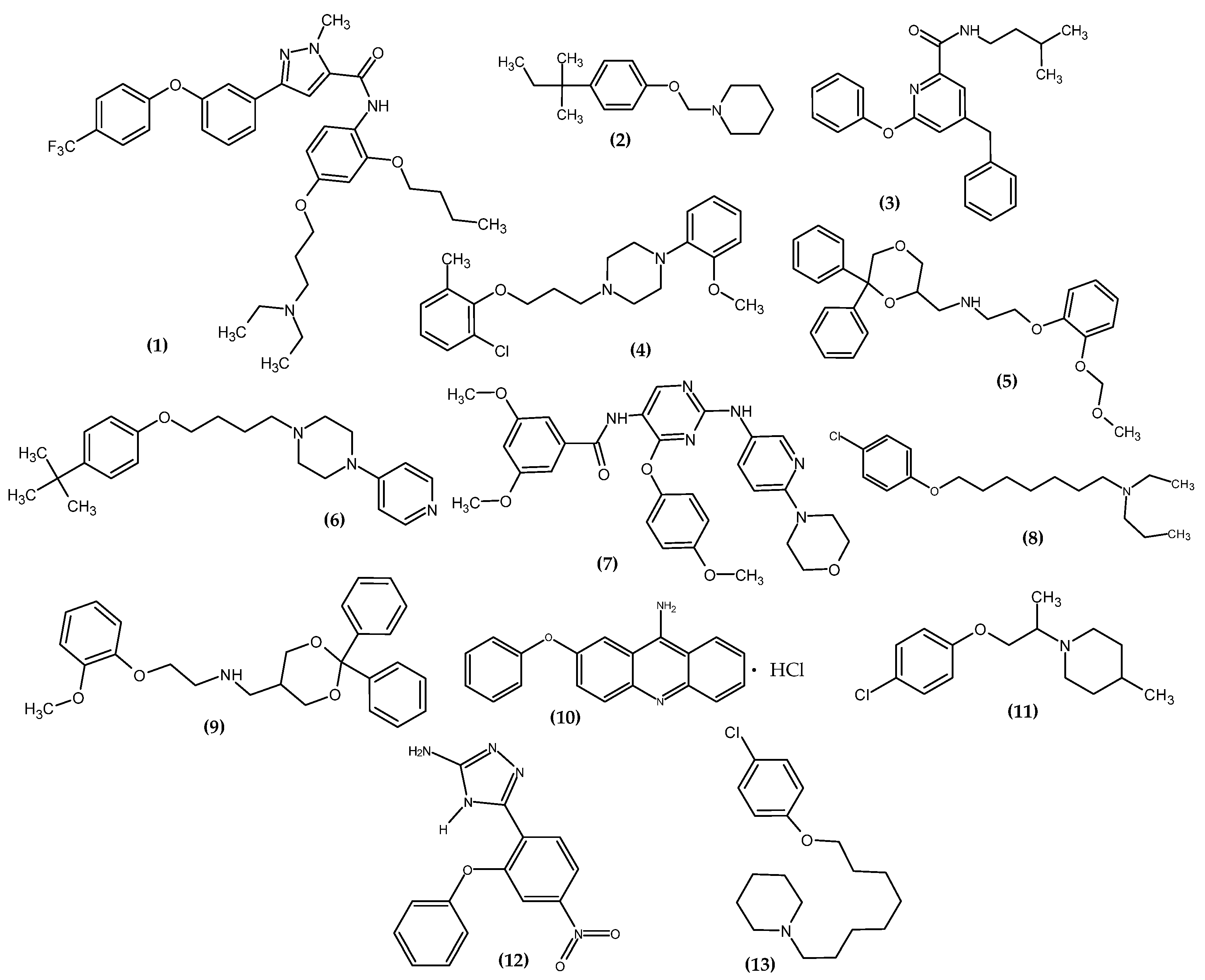
Figure 1. The novel potential agents for a neurological disorder bearing a phenoxy group.
Han et al. obtained pyrazole-5-carboxamides derivatives as potent inhibitors of receptors for advanced glycation end products (RAGE) [3]. RAGE is an inflammatory factor and a critical inducer of oxidative stress, driving the development of Alzheimer’s disease [4]. The most active compound (1) exhibited the highest inhibitory activity against RAGE (83.1 ± 0.5%) with a half-maximal inhibitory concentration (IC50 = 1.9 µM). Studies of directed binding indicated that the modifications improved the binding strength with RAGE, which resulted in an increase in activity. The most active compound bore 4-trifluorophenoxy moiety [3].
Kuder et al. obtained novel tert-amyl phenoxyalkylamine derivatives targeting the histamine H3 receptor (H3R) [5]. H3R is involved in the regulation of the levels of histamine, acetylcholine, serotonin, noradrenaline, glutamate, γ-aminobutyric acid, and neuropeptide Y; therefore, it is a valuable molecular target for potential drugs for Alzheimer’s and Parkinson’s diseases, schizophrenia, narcolepsy, obesity, and the Attention Deficit Hyperactivity Disorder (ADHD) [6][7][8][9][10][11]. Compound (2) showed the highest affinities for human H3R in in vitro binding studies with inhibition constant (Ki = 8.8 nM). In the cAMP accumulation test in the HEK293 cell line, the proposed compound induced a blockade of cAMP level reduction by the receptor agonist—forskolin, which makes it an antagonist of histamine H3R with half maximal effective concentration (EC50 = 157 ± 16 nM). The most active compound bore 4-isopentylphenoxy moiety [5].
Arai et al., based on the Structure–Activity–Relationship (SAR) study of cyclic Aβ16-20 (cyclo-[KLVFF]), identified crucial pharmacophore elements for inhibitors of Aβ aggregation [12]. The KLVFF region is an amino acid peptide fragment of Aβ16-20, related to the formation of Aβ fibrils [13]. Therefore, its inhibition is a potential molecular target in the treatment of Alzheimer’s disease. The authors designed a non-peptide small molecule inhibitor (3) bearing a phenoxy group to play the role of mimicking the phenylalanine side chain (Figure 2). The compound (3) possessed the 71 ± 9.4% of intensity in the thioflavin-T dye assay, related to activity in inhibiting the aggregation of Aβ. The authors stated that the presence of phenoxy group is one of the three, next to the isopentyl carboxamide and benzyl groups, crucial for the activity of this compound [12].
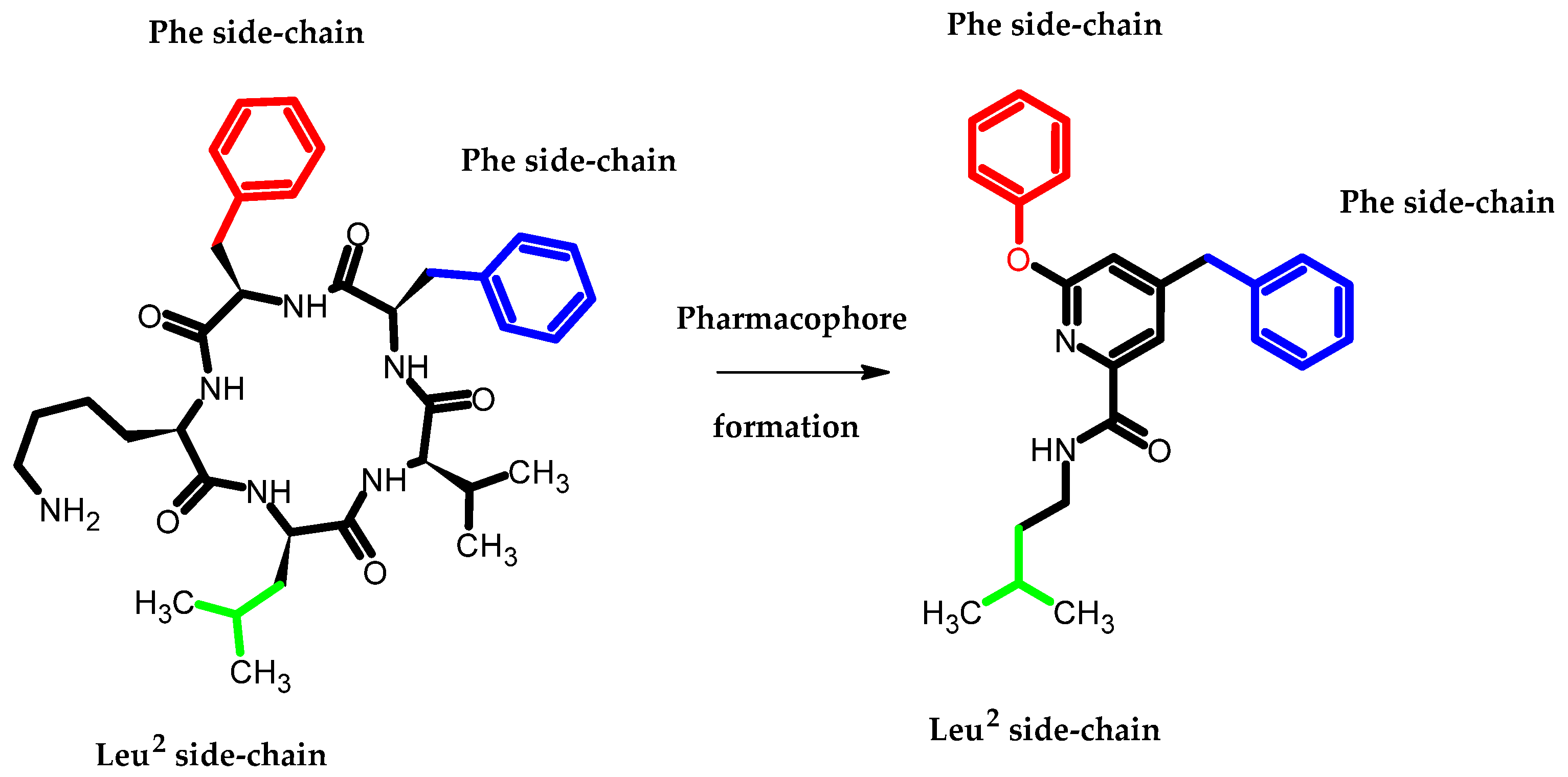
Figure 2. Schematic representation of the developed pharmacophore model in which the terminal phenoxy group plays the role of mimicking the phenylalanine side chain.
Kubacka et al. studied the biological activity of a series of novel aryloxyalkyl derivatives of 2-methoxyphenylpiperazine in a mouse model of Major Depressive Disorder (MDD) [14]. The authors focused on the study of affinity for serotonin receptors, related to the proven role of 5-HT1A, 5-HT2A, 5-HT3, 5-HT6, and 5-HT7 in MDD or anxiety, as well as in antidepressant and anxiolytic effects [15][16][17][18]. They investigated the effect of substituting the phenoxy moiety with chlorine atom and methyl group in various combinations while changing the length of the alkyl chain. Compound (4) exhibited the most exemplary pharmacological properties in the form of partial 5-HT1A receptor agonist, 5-HT2A antagonist and high affinity for 5-HT7, Ki = 0.5 nM, 138.5 nM, and 34 nM, respectively. The results indicated it as an excellent potential antidepressant and anxiolytic agent. The most active compound bore 2-chloro-6-methylphenoxy moiety [14].
Del Bello et al. studied the effect of ortho substitution in the phenoxy moiety and its effect on binding to the 5-HT1A receptor [19]. They designed phenoxyethanamine derivatives and investigated their properties in a mouse model of anxiety in the light/dark exploration test. The methoxymethoxy substituent (5) possessed the best properties. In their research, the authors exhibited that the ortho substitution of the phenoxy group with large substituents increased the proportion of hydrophobic interactions in relation to polar interactions. Moreover, the key conclusion was that the ortho substitution of the phenoxy moiety played the role of structural diversification, enabling the design of compounds that will selectively recognize 5-HT1A over α1d-adrenoceptor and the dopamine D2-like receptor. The phenoxy moiety was responsible for the π–π interactions with Tyr5.39 and Phe6.52, and its ortho substituent stabilized hydrogen bonds with surrounding polar residues and enhanced hydrophobic interactions with the alkyl chains of Val3.33, Ile4.57, and Ile4.59. The engineered derivative acted as a partial agonist of 5-HT1A (binding [35S]GTP γS 5-HT1A pD2 = 9.41) and may be useful in the treatment of disorders related to this receptor. The authors noted the possibility of creating hydrogen bonds with the ether oxygen atom of the phenoxy group. Moreover, they stated that the function of the ether oxygen atom of the phenoxy moiety is presumably favoring the optimal arrangement of the phenyl ring at the binding site and is never involved in direct interactions in complexes with the open model. The most active compound bore 2-methoxy(metoxy)phenoxy substituent [19]. This underlines the privileged nature of the phenoxy moiety as being responsible for improving the interactions with the binding site.
Szczepańska et al. obtained a series of phenoxyalkylamine derivatives and examined their properties in silico, in vitro, and in vivo towards the human H3R (hH3R) [20]. The compound (6) turned out to be the most active, bearing a pyridin-4-yl substituent and a tert-butylphenoxy group linked by a 4-carbon alkyl chain. The novel derivative was characterized as an H3R antagonist. It did not exhibit the highest activity against hH3R in an in vitro study with Ki = 37.8 nM but was the only one in the mouse model to possess anticonvulsant activity in the maximal electroshock-induced seizure (MES). The authors indicated that the ether oxygen atom of the phenoxy moiety was responsible for the formation of hydrogen bonds with TYR3746.51. The most active compound bore the 4-tert-butylphenoxy group [20].
Farag et al. obtained a series of novel dual CSF1R/DAPK1 inhibitors [21]. The structure of new derivatives was designed based on previous research that demonstrated the privilege of the phenoxypyrimidine scaffold for CSF1R inhibitors. Inhibition of colony stimulating factor 1 receptor (CSF1R) ameliorates taupathic neuritis, and inhibition of death-associated protein kinase 1 (DAPK1) inhibits the formation of tau aggregates and exerts neuroprotective properties. A molecular simulation study indicated that the phenoxy moiety was responsible for the interaction with β3 near the hinge region. Compound (7) exhibited the most inhibitory activity of DAPK1 and CSF1R in an in vitro kinase assay with an IC50 = 1.25 µM and 0.15 µM, respectively. The most active compound bore the 4-methoxyphenoxy group [21].
Łażewska et al. obtained chlorophenoxyalkylamine derivatives as potent cholinesterases: acetyl- (AChE) and butyrylcholinesterase (BuChE) [22]. Compound (8) turned out to be the most active against Electrophorus electricus (EeAChE) with an IC50 = 1.93 µM and BuChE from horse serum (EqBuChE) with an IC50 = 1.64 µM. The effect of the chlorine substituent on the phenoxy group is not clear, but it is concluded that the para position slightly increased the activity. The Lineweaver—Burk plot indicated that the proposed compound is a nonselective AChE/BuChE inhibitor. The analysis of the binding mode by docking study exhibited that the phenyl ring of the phenoxy group is responsible for the interactions of π–π with the indole moiety of TRP279 in AChE and the residues of TYR332 in BuChE. The ether oxygen atom of the phenoxy moiety stabilized the inhibitor-AChE complex, and it was related to the formation of hydrogen bonds with the rest of TYR121, a water molecule (1159), and the protonated nitrogen atom. The most active compound bore the 4-chlorophenoxy group [22].
Franchini et al. obtained a series of diphenyl derivatives bearing the phenoxy group with potential 5-HT1A receptor agonist activity [23]. Compound (9) exhibited the highest agonist activity for 5-HT1A receptor with affinity pKi 5-HT1A = 8.8. In rats, the anxiolytic activity was confirmed by the Elevated Plus Maze (EPM) and Open Field tests, and the antidepressive activity was confirmed by the Forced Swim test (Porsolt). Moreover, the antinociceptive activity of the compound was confirmed in the formalin test. The most active compound bore the 2-methoxyphenoxy group [23].
Kaniakova et al. obtained 7-phenoxytacrine (10) [24]. The idea for modifying the tacrine was related to limit its side effects. This resulted in the production of 7-methoxy tacrine, which even reached the 3rd stage of clinical trials, but its further development was halted. This led to the search for further modifications that allowed for the introduction of the phenoxy group. In the Ellman method, the proposed compound exhibited moderate and non-selective potency towards human recombinant AChE, human plasmatic BuChE, and inhibitory activity for GluN1/GluN2B receptors through the ifenprodil binding site with an IC50 (AChE) = 2.4 μM, IC50 (BuChE) = 4.9 μM, IC50 (GluN1/GluN2B) = 1.7 µM, respectively. Most of the N-methyl-D-aspartate receptors (NMDARs) are composed of the GluN1 and GluN2A-D subunits. The authors indicated that this is a unique mechanism of action towards NMDARs relative to previous modifications, indicating a key activity of the phenoxy group. This is confirmed by the results of the docking study, which revealed that the phenoxy moiety is responsible for the preferred π–π interactions with GluN2B (Phe114) and GluN1 (Tyr109) and hydrophobic interactions with GluN1 (Thr110) and GluN2B (Ile111) residues. In the rat model of NMDA-lesion-induced damage, the development of neurodegeneration in the dorsal hippocampus was reduced [24].
Abatematteo et al. obtained novel phenoxyalkylpiperidines as a sigma-1 receptor (σ1R) ligand with potent anti-amnesic activity [25]. σ1R ligands exert antidepressant and antiamnestic activities, and exhibit neuroprotective effects in preclinical models of neurodegenerative diseases [26][27]. Based on previous research, the authors stated that the phenoxy group is one of the crucial chemical groups responsible for hydrophobic interaction for high affinity σ1R [28]. The most active compound (11) bore the 4-chlorophenoxy group and was about 10-fold more active for the σ1R with Ki = 0.34 nM, as compared to the reference standard—Pentazocine with Ki = 3.93 nM [25].
Navidpour et al. obtained novel 4H-1,2,4-triazoles as novel benzodiazepine (BZD) analogues [29]. BZD exerts its action by binding to a specific domain of the GABAA receptor and acts as an anti-drug, a hypnotic, a muscle relaxant, and an anticonvulsant [30][31]. Compound (12) possessed about 100-fold higher affinity for a GABAA/benzodiazepine receptor complex with an IC50 = 0.03 nM, as compared to the reference standard—Diazepam with an IC50 = 2.4 nM. The authors indicated that phenoxy derivatives of a benzodiazepine scaffold are a new class of non-rigid structures with potent anti-seizure activity [29].
Kuder et al. obtained novel chlorophenoxyalkylamine derivatives directed towards H3R [32]. Compound (13) exhibited the highest affinity with Ki = 128 nM and was classified as a cAMP antagonist in a study with HEK293 cells with an EC50 = 75 nM. Moreover, the authors indicated that the position of the ether oxygen atom, i.e., the presence of phenoxy moiety, is of key importance for the high activity of this group of derivatives. It is worth noting that the substitution of the phenoxy group in the para position significantly increased the binding capacity relative to the meta position—in other words, the chlorine atom in the 4 position is the most favored in the case of the H3R binding pocket [32].
2. Anticancer Activity
Cancer is one of the leading causes of death every year [33]. The American Cancer Society predicts that, in the United States alone, more than 1/3 of all newly diagnosed cancer patients will end up dying, which is about 1670 deaths a day [34]. The main factors responsible for the development of neoplasms are environmental factors resulting from human behavior. The most common ones are smoking, excessive alcohol consumption, and diet [35]. The novel potential anticancer agents bearing a phenoxy group are presented in Figure 3.
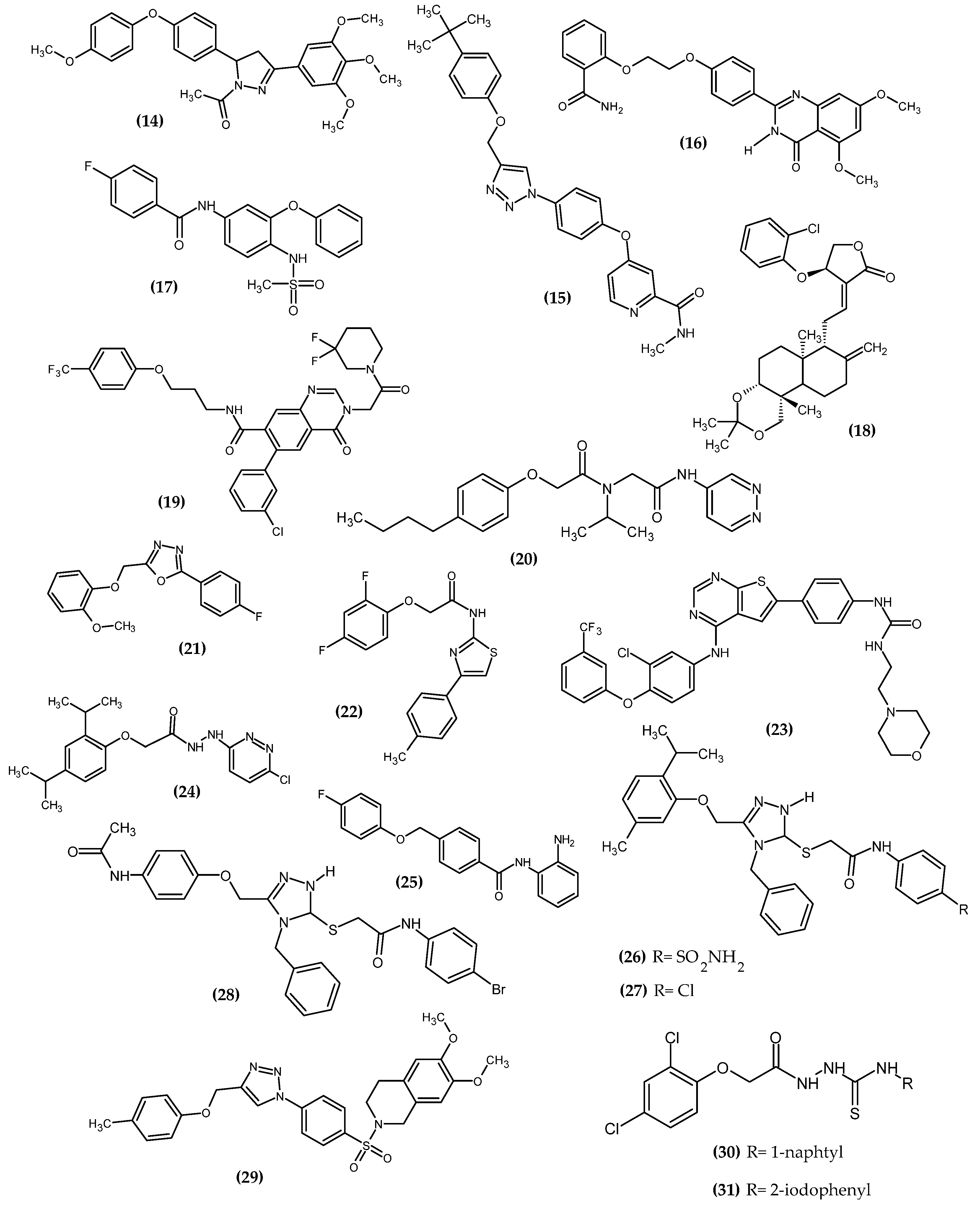
Figure 3. The novel potential agents with anticancer activity bearing a phenoxy group.
Hassan et al. obtained a novel series of pyrazoline derivatives as antiproliferative agents by inhibiting the vascular endothelial growth factor receptor (VEGFR) pathway [36]. Stimulation of the Janus signaling (JAK) pathway and activators of transcriptional proteins 3 (STAT3) in neoplastic cells by hypoxia and inflammatory cytokines results in the secretion of proangiogenic proteins such as vascular endothelial growth factors (VEGF). It activates the phosphorylation of its receptors (VEGFR) in endothelial cells, which leads to the induction of angiogenesis [37][38][39]. Therefore, the search for new potential agents against this signaling pathway allows for controlling tumor proliferation [40]. The basis of the design of the novel substances was the presence of the phenoxy moiety among many drugs approved by the Food and Drug Administration (FDA) as VEGFR inhibitors, e.g., axitinib, lucytanib, and tivozanib. Compound (14) exhibited significant antiproliferative activity against OVCAR-4 ovarian cancer and MDA-MB-468 breast cancer cell lines with an IC50 = 0.29 ± 0.02 μM and 0.35 ± 0.01 μM, respectively, as compared to the reference standard—Staurosporin with an IC50 = 5.86 ± 0.39 µM and 3.45 ± 0.23 µM, respectively. The highest selectivity index (SI = 74) was observed for the MDA-MB-468 breast cancer cell line. The Elisa assay indicated that (14) reduced the VEGF concentration by 85% compared to untreated OVCAR-4 cells. Further studies indicated that the possible mechanism was inhibiting VEGFR-2 phosphorylation by 69.8%, as compared to untreated OVCAR-4 cells and by 77.5%, as compared to untreated MDA-MB-468 cells. The reason for the reduction of VEGF levels may be the inhibition of the transcription factor STAT3 [36], which was confirmed by the docking study. A docking study against STAT3 presented that phenoxyphenyl formed H–π interactions with Glu638, which are crucial for stabilizing the binding conformation (Figure 4). Cell cycle studies exhibited that compound (14) arrested the proliferation of the OVCAR-4 cell cycle in the S phase. It is worth noting that, for other derivatives, one of the modifications was the replacement of the phenoxy with the morpholino group, which resulted in a decrease in the activity of that compound. This underlines the impact of the phenoxy group on activity. The most active compound bore 4-methoxyphenoxy moiety [36].
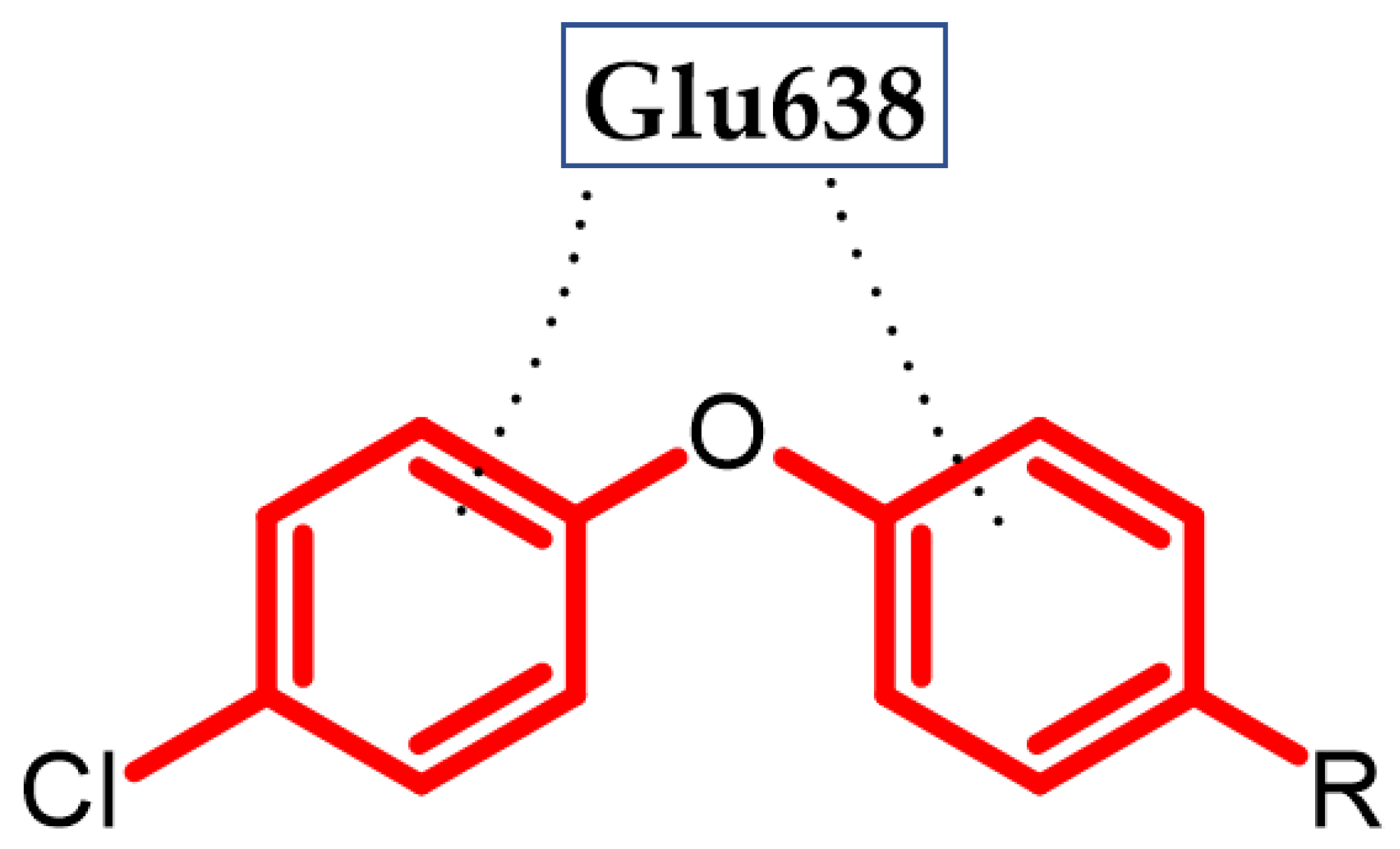
Figure 4. Schematic representation of H-π interactions (black dots) formed by a phenoxyphenyl moiety with STAT3 (R is the rest of the compound structure).
Palakhachane et al. obtained novel Sorafenib analogues bearing the aryl-urea moiety of Sorafenib replaced with a 1,2,3-triazole ring linking the substituted phenoxy fragment [41]. Sorafenib is the first targeted therapy drug for patients with hepatocellular carcinoma. The authors indicated that the terminal phenoxy group could provide a hydrophobic interaction with the lipophilic pocket. Compound (15) was exhibited the highest activity against Huh7 hepatocellular carcinoma cell line with an IC50 = 5.67 ± 0.57 µM. Novel derivative (15) had a higher safety profile against the MRC-5 lung fibroblast cell line, as compared to the reference standard—Sorafenib. The authors noted that the substituted phenoxy group is one of the key structural elements crucial for the selective inhibition of Huh7 with a high safety profile. In other words, the phenoxy group improved the binding affinity and provided greater selectivity and less toxicity of the newer compound. The most active compound bore the tert-butylphenoxy moiety [41].
Chang et al. obtained novel quinazolin-4(3H)-one derivatives bearing phenoxy moiety [42]. The aim of the study was to design a bidirectional inhibitor co-targeting poly (ADP-ribose) polymerase-1 (PARP1) and protein 4-containing bromodomain (BRD4). These two targets reflect the synthetic lethal effect, having cross-links in the global breast cancer network. Compound (16) showed the highest activity against BD1 and BD2 with IC50 = 0.44 μM/L and 0.379 μM/L, respectively. Furthermore, the proposed compound (16) exhibited excellent inhibitory potency against BRD4 with an IC50 = 0.4 μM/L, as compared to the reference standard—RVX-208 with an IC50 = 12.6 μM/L. An antiproliferative activity study for MDA-MB-468 cells for (16) with an IC50 = 3.4–1.1 μM/L was quite lower, as compared to the reference standard—Olaparib with an IC50 = 1.1–1.4 μM/L. Cell cycle study indicated that (16) could interfere with the G1 phase, and could also induce apoptosis in a dose-dependent manner. In the SAR study, the authors stated that the phenoxy group is crucial for high activity, and it is one of the elements of the pharmacophore. The most active compound bore 2-amidophenoxy moiety [42].
Güngor et al. obtained sulfonamide containing Nimesulide derivatives [43]. From the 17th novel compound, the most active (17) bore the 4-fluorophenoxy group. In the sulforhodamine B assay, compound (17) possessed the highest activity against HT-29 colon cancer and the MCF-7 breast cancer cell line with IC50 = 9.24 μM and 11.35 μM, respectively. The mechanism of the antitumor effect was studied by Western blot analyses, which confirmed that expression of pro-apoptotic protein BAX (BCL-2-associated X protein) was upregulated, and anti-apoptotic protein BCL-2 (Bcell lymphoma 2) was downregulated [43].
Li et al. obtained novel andrographolide derivatives in which the 2-chlorophenoxy group was introduced at the atom C14, as a potent antibacterial agent [44]. Further investigations indicated that compound (18) decreased VEGF-induced phosphorylation of Akt, mTOR, MEK1/2, ERK1/2, and p38 MAPK in endothelial cells, and, by inhibiting Akt/mTOR and ERK-dependent pathways, it strongly suppressed tumor cell growth and proliferation. Furthermore (18), by blocking VEGFR-2-mediated signaling, it reduced VEGF expression in tumor cells and inhibited VEGF-induced endothelial cell proliferation, migration, and invasion, which is crucial for anticancer activity [45].
Ma et al. obtain a novel quinazolinone derivative as a cytosolic protein receptor 1 and 2 (NOD1/2) dual antagonist [46]. NOD1 and NOD2 are a key target for immunotherapy due to the presence of a nucleotide-binding oligomerization domain that is an important component of the innate immune system [47][48][49][50][51][52]. Antagonism of both NOD1 and NOD2 signaling guarantees the effectiveness of adjuvant cancer treatment [46]. Compound (19) was not the most active against both NOD1-and NOD2 in HEK293 cells with an IC50 = 1.13 μM and 0.77 μM, respectively; however, it possessed the highest metabolic stability. The authors stated that the oxygen atom as an ether linker connected to the phenoxy group is crucial for high activity. The most active compound bore 4-trifluoromethylphenoxy moiety [46].
Yu et al. obtained novel phenoxy derivatives as non-covalent proteasome inhibitors [53]. The ubiquitin-proteasome system is an important pathway for cell cycle progression [54][55], signal transduction [56], and immune responses [57]; thus, regulation of protease activity by inhibitors is a potential target for cancer therapy. This was confirmed by the FDA approval of bortezomib, carfilzomib, and ixazomib as proteasome inhibitors in anti-cancer therapies [53]. Compound (20) exhibited the highest activity against chymotrypsin-like 20S proteasome. The activity increased with the length of the alkyl chain at the para position of the PhO in the given series: methyl (481 nM) < ethyl (243 nM) < propyl (89 nM) < butyl (49 nM) (20). Positive control—PI-1840 possessed about 2-fold lower activity with IC50 = 92 ± 5 nM. Further lengthening the chain reduced the activity of the compound. The 20S proteasome consists of heptameric rings: two outer α rings and two inner β rings, and each β ring contains three proteolytic subunits β1c, β2c and β5c. Binding mode analysis indicated that the proposed compound possessed selectivity for β5c inhibitory activity with no β5i inhibition, which stated that it could be applied to solid cancers [53].
Lakshmithendral et al. obtained novel 2-(phenoxymethyl)-5-phenyl-1,3,4-oxadiazole derivatives [58]. Compound (21) showed the highest activity against MCF-7 and MDA-MB-453 breast cancer cell lines with an IC50 = 10.51 ± 1.9 μM and IC50 = 10.25 ± 2.5 μM, respectively. Furthermore, the proposed compound induced the apoptosis in the Acridine orange (AO)/ethidium bromide (EtBr) dual staining assay. The most active compound bore the 2-methoxyphenoxyl moiety at the 5-position and the 2-fluorophenyl moiety in the 2-position of the 1,3,4-oxadiazole ring, which determined the anti-cancer activity in the cell proliferation assays according to the authors [58].
Mohammed et al. obtained novel 4-phenyl-2-phenoxyacetamide thiazoles [59]. The trypan blue assay compound (22) possessed the highest activity against both MCF7 and MDA-MB 468 breast cancer cell lines, A549 lung cancer, EAC Ehrlich–Lettre ascites carcinoma, and the DLA Dalton’s lymphoma ascites cell line with IC50 = 14 ± 0.4 μM, 10.2 ± 1 μM, 13.2 ± 0.8 μM, 14. ± 10.2 μM, and 13.9 ± 0.4 μM, respectively. It is worth noting that the MDA-MB-468 cell lines are resistant to chemotherapy related to the lack of biomarkers; however, the proposed compound possessed quite promising activity, while the reference standard—5-Fluorouracil did not exhibit significant cytotoxicity. In the mouse EAC tumor model, the compound (22) reduced tumor growth and extended the life span of the animals. Furthermore, it had no apparent side effects. The most active compound bore 2,4-difluorophenoxy moiety [59].
Milik et al. obtained new thieno [2,3-d] pyrimidine-based dual EGFR/HER2 inhibitors [60]. The epidermal growth factor receptor (EGFR) family belongs to the receptor tyrosine kinases (RTKs) [61] and includes four structurally related RTKs: EGFR (HER1), HER2, HER3, and HER4 [62][63][64]. Dysregulation of EGFR related to mutations or overexpression of receptors results in excessive proliferation [65][66], resistance to apoptosis, and promotes angiogenesis and metastasis [67][68]. EGFR dysregulation is likely associated with non-small cell lung cancer (NSCLC), colorectal cancer, breast cancer, and pancreatic cancer [65][68]; thus, it is a potential target for cancer therapy. Compound (23) was the most active against MDA-MB-361 breast cancer and NCI-H1975 lung adenocarcinoma cancer cell lines with an IC50 = 3.50 ± 0.73 μM and 4.20 ± 0.19 μM, respectively, as compared to the reference standard—Lapatynib with IC50 = 13.73 ± 2.32 μM and 11.46 ± 2.45 μM, respectively. The most active compound bore 3-trifluorophenoxy moiety [60].
Mohammed et al. obtained novel synthesized pyridazine hydrazide appended phenoxy acetic acid [69]. Compound (24) exhibited the highest activity against the A549 lung cancer cell, HepG2 hepatocellular carcinoma cell, A498 kidney cancer cell line, CaSki cellosaurus cell line, and SiHa squamous cell carcinoma with IC50 = 6.6 ± 0.6 μM; 6.9 ± 0.7 μM; 6.8 ± 0.8 μM; 7.5 ± 0.5 μM and 7.8 ± 0.4 μM, respectively, as compared to the reference standard—5-Fluorouracil with IC50 = 7.4 ± 0.5 μM, 8.3 ± 1.8 μM, 5.4 ± 0.7 μM, 7.3 ± 0.4 μM, and 8.3 ± 0.7 μM, respectively. Furthermore, the proposed compound (24) downregulates metalloproteinase 2 (MMP-2) and metalloproteinase 9 (MMP-9) and thereby impaired metastatic cancer cell migration and invasion. The most active compound bore 2,4-diisopropylophenoxy moiety [69].
Xie et al. obtained novel 2-aminobenzamide derivatives [70]. Compound (25) exhibited the highest activity against the HepG2 hepatocellular carcinoma cell line with an IC50 = 3.84 ± 0.54 μM. The possible mechanism of anticancer activity was inducing the G2/M phase cell cycle arrest and apoptosis. Histone deacetylase (HDAC) is a promising target for cancer therapy because it is related to differentiation and apoptosis of cancer cells. Further evaluation showed two times more active potential of the proposed compound against the second isoform of HDAC with IC50 = 0.57 ± 0.09 μM, as compared to the reference standard—CI994 with IC50 = 1.20 ± 0.23 μM [69]. Compound (25) also possessed higher activity against the first isoform of HDAC with IC50 = 1.27 ± 0.20 μM, as compared to the previously mentioned standard with an IC50 = 1.62 ± 0.25 μM; however, its distinctly greater affinity for the second isoform is observed. The most active compound bore 4-fluorophenoxy moiety [70].
Kulabaş et al. obtained the novel 2-(4H-1,2,4-triazole-3-ylthio)acetamide derivatives [71]. Compound (26) exhibited highest activity against the PC-3 prostate cancer cell line, (27) against the A549/ATCC non-small cell lung cancer cell line, and (28) against the K-562 leukemia cancer cell line with IC50 = 5.96 μM, 7.90 μM, and 7.71 μM, respectively. Further studies indicated that (26) triggers apoptosis by using both intrinsic and extrinsic pathways, and (27–28) induce apoptotic cell death by triggering the intrinsic pathway. The most active compounds bore 5-methyl-2-(prop-2-yl)phenoxy and 4-acetylaminophenoxy moiety [71].
Pingaew et al. obtained a novel series of N-benzenesulfonyl-1,2,3,4-tetrahydroisoquinolines [72]. Compound (29) exhibited about 53-fold higher activity with an IC50 = 0.56 ± 0.01 μM against HepG2 hepatocellular carcinoma cell line, as compared to the reference standard—Etoposide with an IC50 = 30.16 ± 0.50. Its activity is even higher than Doxorubicin with an IC50 = 0.79 ± 0.08 μM. Other compounds from these series with phenoxy moiety also exhibited superior inhibitory potency toward HepG2 cells, as compared to the reference standard—Etoposide. The most active compound (29) bore a 6,7-dimethoxy substituent on the isoquinoline core and a p-tolyl group on the triazole moiety [72].
Gupta et al. obtained novel phenoxy thiosemicarbazide derivatives as potent antibacterial and insecticidal agent [73]. Virtual screening was the basis for testing the anti-cancer potential. The proposed compound induced apoptosis by increasing the cell population in either S-phase or G2-phase. Molecular docking has shown that (30) acted as a DNA intercalator. The most active compound bore 1-naphtyl substituent [74]. One last studies of 2,4-dichlorophenoxy hydrazide derivatives (31) revealed anti-melanoma activity on the G-361 melanoma cell line with an IC50 = 112 ± 4.76 μM [75]. The proposed compound was not the most active, but it exhibited a safety profile for the normal fibroblast. The possible mechanism of action downregulated the expression of dihydroorotate dehydrogenase (DHODH), which is crucial for nucleotide synthesis. Given the upregulated nature of this process as a result of proliferation, DHODH inhibitors represent a new hope for targeted therapy for melanoma [76]. One review of the literature on the latest anti-melanoma agents revealed that the phenoxy derivatives are among the first in recent years [77]. The most active compound bore a 2-iodophenyl substituent [75].
2.1 Bruton Tyrosine Kinase Inhibitors
Bruton Tyrosine Kinase (BTK) plays a key role in, among others, B-cell antigen receptor (BCR) signal transduction in normal and malignant B lymphocytes. BKT inhibitors are a new approach in the chemotherapy of chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) [78]. The novel potential BTK inhibitors bearing a phenoxy group are presented in Figure 5.
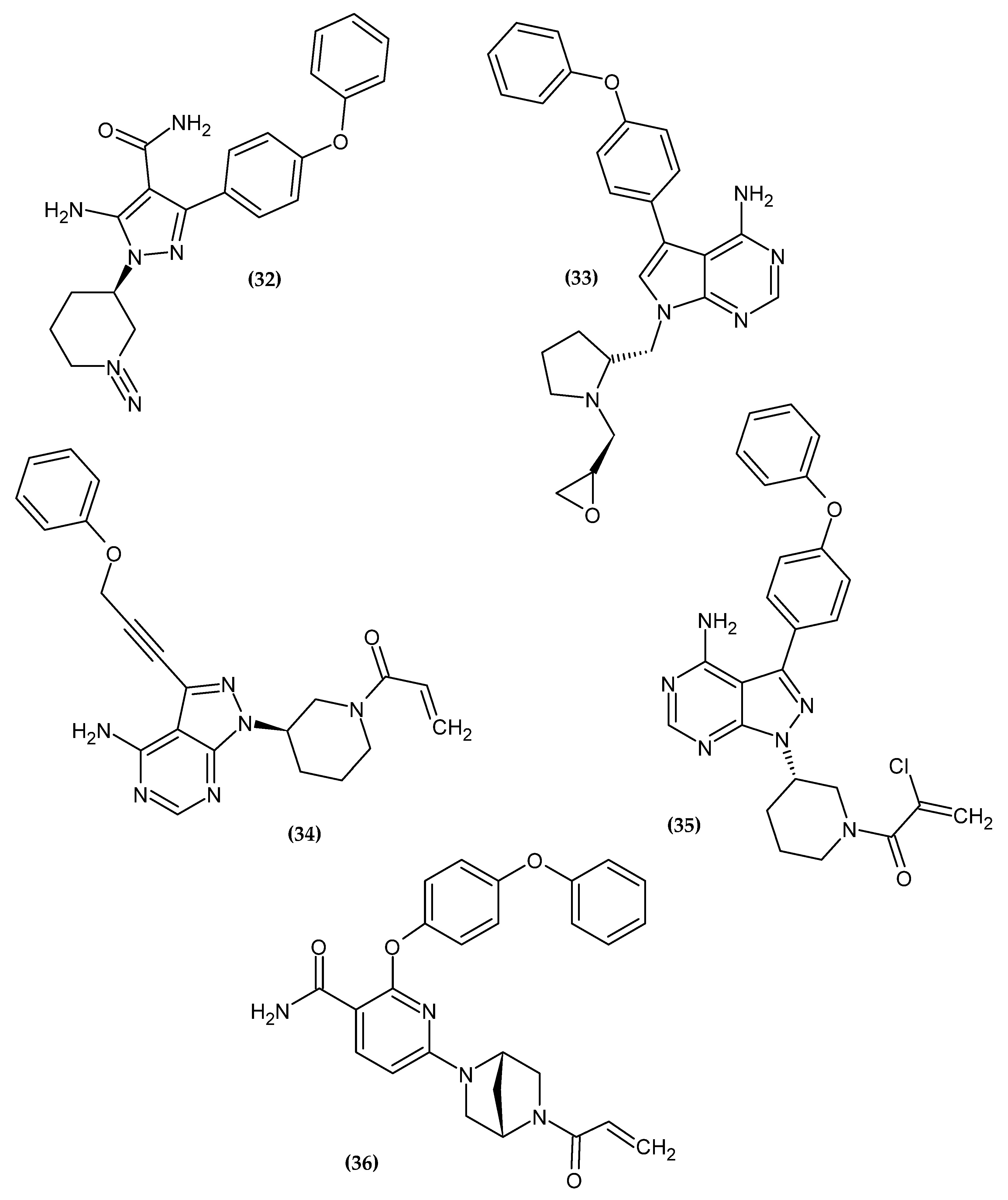
Figure 5. The novel potential BTK inhibitors bearing a phenoxy group.
Schnute et al. obtained a novel aminopyrazole carboxamide as a potent Bruton’s Tyrosine Kinase Inhibitor [79]. Compound (32) exhibited the best inhibitory activity against both wild type BTK and Cys481S BTK with an IC50 = 0.37 nM and 2.8 nM, respectively. Derivatives bearing unsubstituted phenoxy moiety had the highest activity [79].
Zhang et al. obtained novel 7H-pyrrolo [2,3-d]pyrimidin-4-amine derivatives as novel anti-arthritic agents [80]. Compound (33) exhibited the most excellent potency against Ramos and Jeko-1, the B-cell lymphoma cell lines, and Daudi BTK enhanced cell line with IC50 = 8.52 μM, 11.10 μM, and 7.04 μM, respectively. Furthermore, in an enzymatic assay, it possessed the highest inhibitory potential for BTK with an IC50 = 3.0 nM. Molecular docking revealed that phenoxy moiety is responsible for hydrophobic interaction [80].
Zheng et al. focused on modifying the phenyl chain linking the phenoxy group to the pyrazolopyrimidine core of Ibrutinib [81]. Modification of the elongation of this chain improves the phenoxy interaction. Compound (34) exhibited excellent potency Ramos and Raji for the B-cell lymphoma cell lines with an IC50 = 8.91 μM and 1.80 μM, respectively. Furthermore, in an enzymatic assay, it had the highest inhibitory potential for BTK with an IC50 = 7.95 nM. Terminal phenoxy moiety is responsible for pi-stacking interaction [81] (Figure 6).
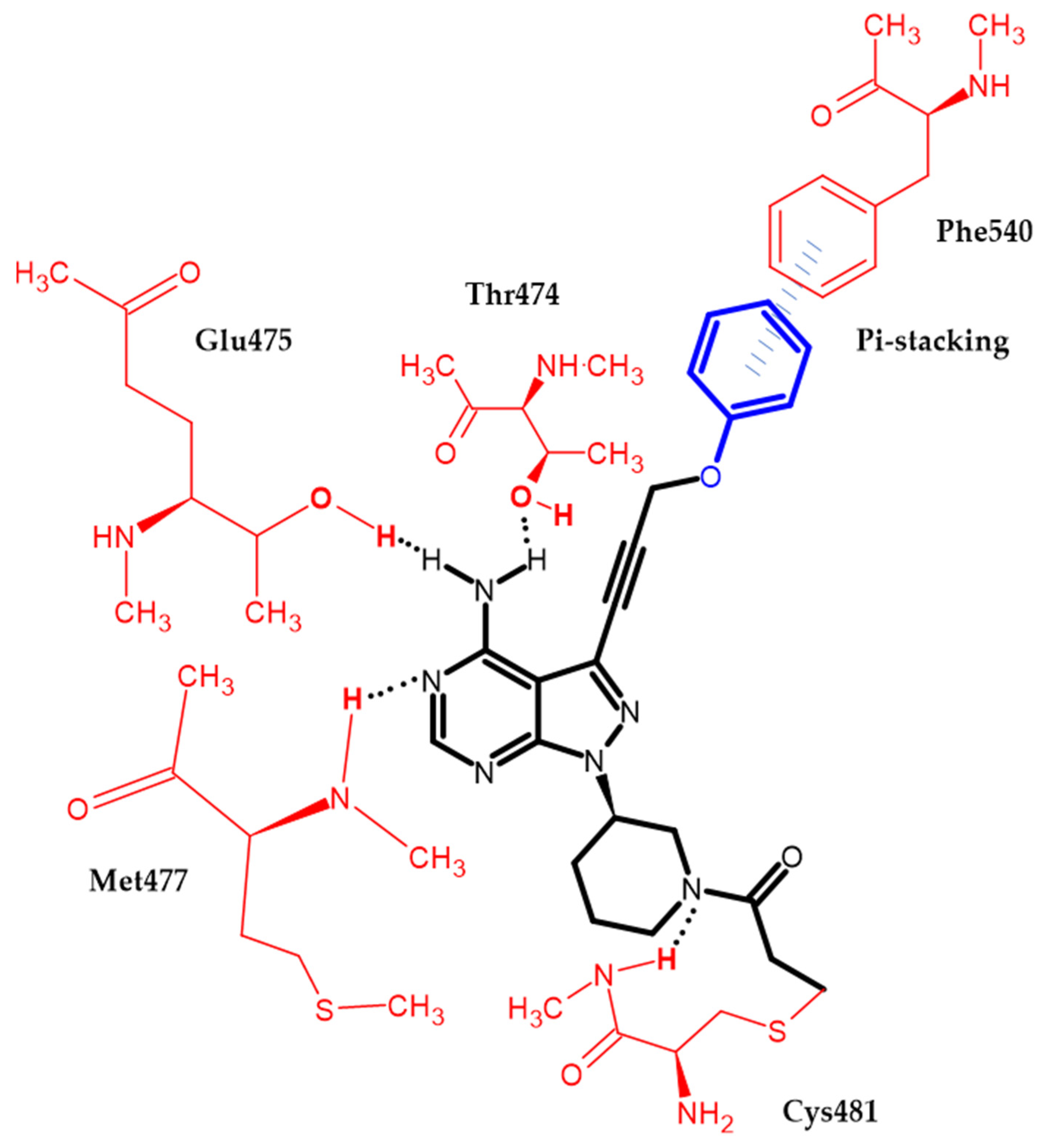
Figure 6. Schematic representation of the pi-stacking (blue lines) interaction formed by a phenoxy group with a BTK enzyme. The black dots represent hydrogen bonds.
Huang et al. showed another analog with a chlorine atom attached to the acrylamide head of Ibrutinib [82]. The proposed compound (35) had high activity against recombinant human BTK kinase with an IC50 = 2.5 nM. Furthermore, it also exhibited potent inhibitory activity against LY-10, DOHH-2, REC-1, and Mino lymphoma cell lines with IC50 = 0.16 μM, 0.22 μM, 0.01 μM and 0.56 μM, respectively [82].
Qiu et al. obtained a novel irreversible covalent BTK inhibitor [83]. The authors initially introduced a morpholinocarbonylphenoxy substituent into the pyridine backbone replacing the morpholinocarbonylphenylamine substituent, which resulted in a significant loss of activity, possibly due to a flip of the O-linked group into the selectivity pocket. Another modification was the introduction of a phenoxyphenoxy substituent, which resulted in a 25-fold loss of potency but significantly improved the permeability profile. The introduction of a phenoxyphenoxy group is crucial for the interaction with the selectivity pocket of the BTK kinase domain [83][84][85][86][87]. It is used as a selectivity pocket group. Knowing the beneficial effect of the phenoxyphenoxy substituent, the authors focused on modifying the linker between the core pyrimidine and ethenylcarbonyl. Compound (36) exhibited the best inhibitory potency in an enzymatic assay against BTK with an IC50 = 0.7 nM. A molecular docking study revealed that the phenoxyphenoxy group produced hydrogen bonds with Lys430 and Asp539 and occupied a selective pocket. This is another example of an active compound bearing phenoxyphenoxy moiety [83].
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480.
- Cui, L.; Hou, N.-N.; Wu, H.-M.; Zuo, X.; Lian, Y.-Z.; Zhang, C.-N.; Wang, Z.-F.; Zhang, X.; Zhu, J.-H. Prevalence of Alzheimer’s Disease and Parkinson’s Disease in China: An Updated Systematical Analysis. Front. Aging Neurosci. 2020, 12, 603854.
- Han, Y.T.; Kim, K.; Choi, G.-I.; An, H.; Son, D.; Kim, H.; Ha, H.-J.; Son, J.-H.; Chung, S.-J.; Park, H.-J.; et al. Pyrazole-5-Carboxamides, Novel Inhibitors of Receptor for Advanced Glycation End Products (RAGE). Eur. J. Med. Chem. 2014, 79, 128–142.
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495.
- Kuder, K.J.; Łażewska, D.; Kaleta, M.; Latacz, G.; Kottke, T.; Olejarz, A.; Karcz, T.; Fruziński, A.; Szczepańska, K.; Karolak-Wojciechowska, J.; et al. Synthesis and Biological Activity of Novel Tert-Amylphenoxyalkyl (Homo)Piperidine Derivatives as Histamine H3R Ligands. Bioorg. Med. Chem. 2017, 25, 2701–2712.
- Tiligada, E.; Kyriakidis, K.; Chazot, P.L.; Passani, M.B. Histamine Pharmacology and New CNS Drug Targets. CNS Neurosci. Ther. 2011, 17, 620–628.
- Walter, M.; Stark, H. Histamine Receptor Subtypes: A Century of Rational Drug Design. Front. Biosci. 2012, 4, 461–488.
- Schlicker, E.; Kathmann, M. Modulation of In Vitro Neurotransmission in the CNS and in the Retina via H3 Heteroreceptors. In Pharmacochemistry Library; Leurs, R., Timmerman, H., Eds.; The Histamine H Receptor; Elsevier: Amsterdam, The Netherlands, 1998; Volume 30, pp. 13–26.
- Blandina, P.; Bacciottini, L.; Giovannini, M.G.; Mannaioni, P.F. H3 Receptor Modulation of the Release of Neurotransmitters In Vivo. In Pharmacochemistry Library; Leurs, R., Timmerman, H., Eds.; The Histamine H Receptor; Elsevier: Amsterdam, The Netherlands, 1998; Volume 30, pp. 27–40.
- Tiligada, E.; Zampeli, E.; Sander, K.; Stark, H. Histamine H3 and H4 Receptors as Novel Drug Targets. Expert Opin. Investig. Drugs 2009, 18, 1519–1531.
- Berlin, M.; Boyce, C.W.; de Lera Ruiz, M. Histamine H3 Receptor as a Drug Discovery Target. J. Med. Chem. 2011, 54, 26–53.
- Arai, T.; Araya, T.; Sasaki, D.; Taniguchi, A.; Sato, T.; Sohma, Y.; Kanai, M. Rational Design and Identification of a Non-Peptidic Aggregation Inhibitor of Amyloid-β Based on a Pharmacophore Motif Obtained from Cyclo. Angew. Chem. Int. Ed. 2014, 53, 8236–8239.
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747.
- Kubacka, M.; Mogilski, S.; Bednarski, M.; Nowiński, L.; Dudek, M.; Żmudzka, E.; Siwek, A.; Waszkielewicz, A.M.; Marona, H.; Satała, G.; et al. Antidepressant-like Activity of Aroxyalkyl Derivatives of 2-Methoxyphenylpiperazine and Evidence for the Involvement of Serotonin Receptor Subtypes in Their Mechanism of Action. Pharmacol. Biochem. Behav. 2016, 141, 28–41.
- Carr, G.V.; Schechter, L.E.; Lucki, I. Antidepressant and Anxiolytic Effects of Selective 5-HT6 Receptor Agonists in Rats. Psychopharmacology 2011, 213, 499–507.
- Artigas, F. Serotonin Receptors Involved in Antidepressant Effects. Pharmacol. Ther. 2013, 137, 119–131.
- Fakhoury, M. Revisiting the Serotonin Hypothesis: Implications for Major Depressive Disorders. Mol. Neurobiol. 2016, 53, 2778–2786.
- Waszkielewicz, A.M.; Pytka, K.; Rapacz, A.; Wełna, E.; Jarzyna, M.; Satała, G.; Bojarski, A.; Sapa, J.; Żmudzki, P.; Filipek, B.; et al. Synthesis and Evaluation of Antidepressant-like Activity of Some 4-Substituted 1-(2-Methoxyphenyl)Piperazine Derivatives. Chem. Biol. Drug Des. 2015, 85, 326–335.
- Del Bello, F.; Bonifazi, A.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Petrelli, R.; Cifani, C.; Micioni Di Bonaventura, M.V.; Keck, T.M.; Mazzolari, A.; et al. The Replacement of the 2-Methoxy Substituent of N-((6,6-Diphenyl-1,4-Dioxan-2-Yl)Methyl)-2-(2-Methoxyphenoxy)Ethan-1-Amine Improves the Selectivity for 5-HT1A Receptor over A1-Adrenoceptor and D2-like Receptor Subtypes. Eur. J. Med. Chem. 2017, 125, 233–244.
- Szczepańska, K.; Karcz, T.; Mogilski, S.; Siwek, A.; Kuder, K.J.; Latacz, G.; Kubacka, M.; Hagenow, S.; Lubelska, A.; Olejarz, A.; et al. Synthesis and Biological Activity of Novel Tert-Butyl and Tert-Pentylphenoxyalkyl Piperazine Derivatives as Histamine H3R Ligands. Eur. J. Med. Chem. 2018, 152, 223–234.
- Farag, A.K.; Hassan, A.H.E.; Jeong, H.; Kwon, Y.; Choi, J.G.; Oh, M.S.; Park, K.D.; Kim, Y.K.; Roh, E.J. First-in-Class DAPK1/CSF1R Dual Inhibitors: Discovery of 3,5-Dimethoxy-N-(4-(4-Methoxyphenoxy)-2-((6-Morpholinopyridin-3-Yl)Amino)Pyrimidin-5-Yl)Benzamide as a Potential Anti-Tauopathies Agent. Eur. J. Med. Chem. 2019, 162, 161–175.
- Franchini, S.; Sorbi, C.; Linciano, P.; Carnevale, G.; Tait, A.; Ronsisvalle, S.; Buccioni, M.; Del Bello, F.; Cilia, A.; Pirona, L.; et al. 1,3-Dioxane as a Scaffold for Potent and Selective 5-HT1AR Agonist with in-Vivo Anxiolytic, Anti-Depressant and Anti-Nociceptive Activity. Eur. J. Med. Chem. 2019, 176, 310–325.
- Łażewska, D.; Jończyk, J.; Bajda, M.; Szałaj, N.; Więckowska, A.; Panek, D.; Moore, C.; Kuder, K.; Malawska, B.; Kieć-Kononowicz, K. Cholinesterase Inhibitory Activity of Chlorophenoxy Derivatives—Histamine H3 Receptor Ligands. Bioorg. Med. Chem. Lett. 2016, 26, 4140–4145.
- Kaniakova, M.; Korabecny, J.; Holubova, K.; Kleteckova, L.; Chvojkova, M.; Hakenova, K.; Prchal, L.; Novak, M.; Dolezal, R.; Hepnarova, V.; et al. 7-Phenoxytacrine Is a Dually Acting Drug with Neuroprotective Efficacy In Vivo. Biochem. Pharmacol. 2021, 186, 114460.
- Abatematteo, F.S.; Mosier, P.D.; Niso, M.; Brunetti, L.; Berardi, F.; Loiodice, F.; Contino, M.; Delprat, B.; Maurice, T.; Laghezza, A.; et al. Development of Novel Phenoxyalkylpiperidines as High-Affinity Sigma-1 (Σ1) Receptor Ligands with Potent Anti-Amnesic Effect. Eur. J. Med. Chem. 2022, 228, 114038.
- Maurice, T.; Goguadze, N. Role of Σ1 Receptors in Learning and Memory and Alzheimer’s Disease-Type Dementia. Adv. Exp. Med. Biol. 2017, 964, 213–233.
- Maurice, T.; Goguadze, N. Sigma-1 (Σ1) Receptor in Memory and Neurodegenerative Diseases. In Sigma Proteins: Evolution of the Concept of Sigma Receptors; Kim, F.J., Pasternak, G.W., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2017; pp. 81–108. ISBN 978-3-319-65853-7.
- Berardi, F.; Ferorelli, S.; Abate, C.; Pedone, M.P.; Colabufo, N.A.; Contino, M.; Perrone, R. Methyl Substitution on the Piperidine Ring of N- Derivatives as a Probe for Selective Binding and Activity at the Σ1 Receptor. J. Med. Chem. 2005, 48, 8237–8244.
- Navidpour, L.; Shabani, S.; Heidari, A.; Bashiri, M.; Ebrahim-Habibi, A.; Shahhosseini, S.; Shafaroodi, H.; Abbas Tabatabai, S.; Toolabi, M. 5--4H-1,2,4-Triazoles as Novel Flexible Benzodiazepine Analogues: Synthesis, Receptor Binding Affinity and Lipophilicity-Dependent Anti-Seizure Onset of Action. Bioorg. Chem. 2021, 106, 104504.
- Sternbach, L.H. The Benzodiazepine Story. J. Med. Chem. 1979, 22, 1–7.
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA Receptor Signalling Mechanisms Revealed by Structural Pharmacology. Nature 2019, 565, 454–459.
- Kuder, K.; Łażewska, D.; Latacz, G.; Schwed, J.S.; Karcz, T.; Stark, H.; Karolak-Wojciechowska, J.; Kieć-Kononowicz, K. Chlorophenoxy Aminoalkyl Derivatives as Histamine H3R Ligands and Antiseizure Agents. Bioorg. Med. Chem. 2016, 24, 53–72.
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27.
- 2022 Cancer Facts & Figures Cancer|Cancer Death Rate Drops. Available online: https://www.cancer.org/latest-news/facts-and-figures-2022.html (accessed on 29 March 2022).
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental Risk Factors for Cancer-Review Paper. Ann Agric Environ Med. 2019, 26, 1–7.
- Hassan, R.A.; Emam, S.H.; Hwang, D.; Kim, G.-D.; Hassanin, S.O.; Khalil, M.G.; Abdou, A.M.; Sonousi, A. Design, Synthesis and Evaluation of Anticancer Activity of New Pyrazoline Derivatives by down-Regulation of VEGF: Molecular Docking and Apoptosis Inducing Activity. Bioorg. Chem. 2022, 118, 105487.
- Glade-Bender, J.; Kandel, J.J.; Yamashiro, D.J. VEGF Blocking Therapy in the Treatment of Cancer. Expert Opin. Biol. Ther. 2003, 3, 263–276.
- Prager, G.W.; Poettler, M.; Unseld, M.; Zielinski, C.C. Angiogenesis in Cancer: Anti-VEGF Escape Mechanisms. Transl. Lung Cancer Res. 2012, 1, 14–25.
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 Activity Up-Regulates VEGF Expression and Tumor Angiogenesis. Oncogene 2002, 21, 2000–2008.
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical Antiangiogenesis and Antitumor Activities of Axitinib (AG-013736), an Oral, Potent, and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283.
- Palakhachane, S.; Ketkaew, Y.; Chuaypen, N.; Sirirak, J.; Boonsombat, J.; Ruchirawat, S.; Tangkijvanich, P.; Suksamrarn, A.; Limpachayaporn, P. Synthesis of Sorafenib Analogues Incorporating a 1,2,3-Triazole Ring and Cytotoxicity towards Hepatocellular Carcinoma Cell Lines. Bioorg. Chem. 2021, 112, 104831.
- Chang, X.; Sun, D.; Shi, D.; Wang, G.; Chen, Y.; Zhang, K.; Tan, H.; Liu, J.; Liu, B.; Ouyang, L. Design, Synthesis, and Biological Evaluation of Quinazolin-4(3H)-One Derivatives Co-Targeting Poly(ADP-Ribose) Polymerase-1 and Bromodomain Containing Protein 4 for Breast Cancer Therapy. Acta Pharm. Sin. B 2021, 11, 156–180.
- Güngör, T.; Ozleyen, A.; Yılmaz, Y.B.; Siyah, P.; Ay, M.; Durdağı, S.; Tumer, T.B. New Nimesulide Derivatives with Amide/Sulfonamide Moieties: Selective COX-2 Inhibition and Antitumor Effects. Eur. J. Med. Chem. 2021, 221, 113566.
- Li, F.; Li, X.-M.; Sheng, D.; Chen, S.-R.; Nie, X.; Liu, Z.; Wang, D.; Zhao, Q.; Wang, Y.; Wang, Y.; et al. Discovery and Preliminary SAR of 14-Aryloxy-Andrographolide Derivatives as Antibacterial Agents with Immunosuppressant Activity. RSC Adv. 2018, 8, 9440–9456.
- Li, J.; Li, F.; Tang, F.; Zhang, J.; Li, R.; Sheng, D.; Lee, S.M.-Y.; Zhou, G.-C.; Leung, G.P.-H. AGS-30, an Andrographolide Derivative, Suppresses Tumor Angiogenesis and Growth in Vitro and in Vivo. Biochem. Pharmacol. 2020, 171, 113694.
- Ma, Y.; Yang, J.; Wei, X.; Pei, Y.; Ye, J.; Li, X.; Si, G.; Tian, J.; Dong, Y.; Liu, G. Nonpeptidic Quinazolinone Derivatives as Dual Nucleotide-Binding Oligomerization Domain-like Receptor 1/2 Antagonists for Adjuvant Cancer Chemotherapy. Eur. J. Med. Chem. 2020, 207, 112723.
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like Proteins in Immunity, Inflammation and Disease. Nat. Immunol. 2006, 7, 1250–1257.
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908.
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in Innate Immunity and Inflammatory Diseases. Biosci. Rep. 2012, 32, 597–608.
- Miceli-Richard, C.; Lesage, S.; Rybojad, M.; Prieur, A.M.; Manouvrier-Hanu, S.; Häfner, R.; Chamaillard, M.; Zouali, H.; Thomas, G.; Hugot, J.P. CARD15 Mutations in Blau Syndrome. Nat. Genet. 2001, 29, 19–20.
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A Frameshift Mutation in NOD2 Associated with Susceptibility to Crohn’s Disease. Nature 2001, 411, 603–606.
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327.
- Yu, J.; Xu, L.; Hong, D.; Zhang, X.; Liu, J.; Li, D.; Li, J.; Zhou, Y.; Liu, T. Design, Synthesis, and Biological Evaluation of Novel Phenol Ether Derivatives as Non-Covalent Proteasome Inhibitors. Eur. J. Med. Chem. 2019, 161, 543–558.
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How Proteolysis Drives the Cell Cycle. Science 1996, 274, 1652–1659.
- Ciechanover, A. The Ubiquitin-Proteasome Pathway: On Protein Death and Cell Life. EMBO J. 1998, 17, 7151–7160.
- Chen, J.-J.; Lin, F.; Qin, Z.-H. The Roles of the Proteasome Pathway in Signal Transduction and Neurodegenerative Diseases. Neurosci. Bull. 2008, 24, 183–194.
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A Selective Inhibitor of the Immunoproteasome Subunit LMP7 Blocks Cytokine Production and Attenuates Progression of Experimental Arthritis. Nat. Med. 2009, 15, 781–787.
- Lakshmithendral, K.; Saravanan, K.; Elancheran, R.; Archana, K.; Manikandan, N.; Arjun, H.A.; Ramanathan, M.; Lokanath, N.K.; Kabilan, S. Design, Synthesis and Biological Evaluation of 2-(Phenoxymethyl)-5-Phenyl-1,3,4-Oxadiazole Derivatives as Anti-Breast Cancer Agents. Eur. J. Med. Chem. 2019, 168, 1–10.
- Mohammed, Y.H.E.; Malojirao, V.H.; Thirusangu, P.; Al-Ghorbani, M.; Prabhakar, B.T.; Khanum, S.A. The Novel 4-Phenyl-2-Phenoxyacetamide Thiazoles Modulates the Tumor Hypoxia Leading to the Crackdown of Neoangiogenesis and Evoking the Cell Death. Eur. J. Med. Chem. 2018, 143, 1826–1839.
- Milik, S.N.; Abdel-Aziz, A.K.; Lasheen, D.S.; Serya, R.A.T.; Minucci, S.; Abouzid, K.A.M. Surmounting the Resistance against EGFR Inhibitors through the Development of Thieno Pyrimidine-Based Dual EGFR/HER2 Inhibitors. Eur. J. Med. Chem. 2018, 155, 316–336.
- Blume-Jensen, P.; Hunter, T. Oncogenic Kinase Signalling. Nature 2001, 411, 355–365.
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174.
- Citri, A.; Yarden, Y. EGF–ERBB Signalling: Towards the Systems Level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516.
- Hynes, N.E.; Lane, H.A. ERBB Receptors and Cancer: The Complexity of Targeted Inhibitors. Nat. Rev. Cancer 2005, 5, 341–354.
- Rowinsky, E.K. The ErbB Family: Targets for Therapeutic Development against Cancer and Therapeutic Strategies Using Monoclonal Antibodies and Tyrosine Kinase Inhibitors. Annu. Rev. Med. 2004, 55, 433–457.
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor-Tyrosine Kinases. Cell 2010, 141, 1117–1134.
- Bianco, R.; Gelardi, T.; Damiano, V.; Ciardiello, F.; Tortora, G. Rational Bases for the Development of EGFR Inhibitors for Cancer Treatment. Int. J. Biochem. Cell Biology 2007, 39, 1416–1431.
- Kamath, S.; Buolamwini, J.K. Targeting EGFR and HER-2 Receptor Tyrosine Kinases for Cancer Drug Discovery and Development. Med. Res. Rev. 2006, 26, 569–594.
- Mohammed, Y.H.E.; Thirusangu, P.; Zabiulla; Vigneshwaran, V.; Prabhakar, B.T.; Khanum, S.A. The Anti-Invasive Role of Novel Synthesized Pyridazine Hydrazide Appended Phenoxy Acetic Acid against Neoplastic Development Targeting Matrix Metallo Proteases. Biomed. Pharmacother. 2017, 95, 375–386.
- Xie, R.; Yao, Y.; Tang, P.; Chen, G.; Liu, X.; Yun, F.; Cheng, C.; Wu, X.; Yuan, Q. Design, Synthesis and Biological Evaluation of Novel Hydroxamates and 2-Aminobenzamides as Potent Histone Deacetylase Inhibitors and Antitumor Agents. Eur. J. Med. Chem. 2017, 134, 1–12.
- Kulabaş, N.; Tatar, E.; Bingöl Özakpınar, Ö.; Özsavcı, D.; Pannecouque, C.; De Clercq, E.; Küçükgüzel, İ. Synthesis and Antiproliferative Evaluation of Novel 2-(4H-1,2,4-Triazole-3-Ylthio)Acetamide Derivatives as Inducers of Apoptosis in Cancer Cells. Eur. J. Med. Chem. 2016, 121, 58–70.
- Pingaew, R.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Design, Synthesis and Molecular Docking Studies of Novel N-Benzenesulfonyl-1,2,3,4-Tetrahydroisoquinoline-Based Triazoles with Potential Anticancer Activity. Eur. J. Med. Chem. 2014, 81, 192–203.
- Sen Gupta, A.K.; Misra, H.K. Studies on potential pesticides. Part XIII: Synthesis and evaluation of S-(3-substituted phenoxymethyl-4-aryl/ayclohexyl-4H-1,2,4-triazol-5-yl)-2-mercaptomethylbenzimidazo-les for antibacterial and insecticidal activities. J. Indian Chem. Soc. 1981, 58, 508–511.
- Pitucha, M.; Korga-Plewko, A.; Kozyra, P.; Iwan, M.; Kaczor, A.A. 2,4-Dichlorophenoxyacetic Thiosemicarbazides as a New Class of Compounds against Stomach Cancer Potentially Intercalating with DNA. Biomolecules 2020, 10, 296.
- Kozyra, P.; Korga-Plewko, A.; Karczmarzyk, Z.; Hawrył, A.; Wysocki, W.; Człapski, M.; Iwan, M.; Ostrowska-Leśko, M.; Fornal, E.; Pitucha, M. Potential Anticancer Agents against Melanoma Cells Based on an As-Synthesized Thiosemicarbazide Derivative. Biomolecules 2022, 12, 151.
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and Cancer: Promising Prospects to Be Explored. Cancer Metab. 2021, 9, 22.
- Kozyra, P.; Krasowska, D.; Pitucha, M. New Potential Agents for Malignant Melanoma Treatment—Most Recent Studies 2020–2022. Int. J. Mol. Sci. 2022, 23, 6084.
- Burger, J.A. BTK Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393.
- Schnute, M.E.; Benoit, S.E.; Buchler, I.P.; Caspers, N.; Grapperhaus, M.L.; Han, S.; Hotchandani, R.; Huang, N.; Hughes, R.O.; Juba, B.M.; et al. Aminopyrazole Carboxamide Bruton’s Tyrosine Kinase Inhibitors. Irreversible to Reversible Covalent Reactive Group Tuning. ACS Med. Chem. Lett. 2019, 10, 80–85.
- Zhang, C.; Pei, H.; He, J.; Zhu, J.; Li, W.; Niu, T.; Xiang, M.; Chen, L. Design, Synthesis and Evaluation of Novel 7H-PyrroloPyrimidin-4-Amine Derivatives as Potent, Selective and Reversible Bruton’s Tyrosine Kinase (BTK) Inhibitors for the Treatment of Rheumatoid Arthritis. Eur. J. Med. Chem. 2019, 169, 121–143.
- Zheng, N.; Pan, J.; Hao, Q.; Li, Y.; Zhou, W. Design, Synthesis and Biological Evaluation of Novel 3-Substituted Pyrazolopyrimidine Derivatives as Potent Bruton’s Tyrosine Kinase (BTK) Inhibitors. Bioorg. Med. Chem. 2018, 26, 2165–2172.
- Huang, W.; Wang, S.; Zhang, Z.; Zhang, C.; Zeng, S.; Liang, M.; Shen, Z.; Liu, X. HZ-A-005, a Potent, Selective, and Covalent Bruton’s Tyrosine Kinase Inhibitor in Preclinical Development. Bioorg. Chem. 2020, 105, 104377.
- Qiu, H.; Liu-Bujalski, L.; Caldwell, R.D.; Follis, A.V.; Gardberg, A.; Goutopoulos, A.; Grenningloh, R.; Head, J.; Johnson, T.; Jones, R.; et al. Discovery of Potent, Highly Selective Covalent Irreversible BTK Inhibitors from a Fragment Hit. Bioorg. Med. Chem. Lett. 2018, 28, 2939–2944.
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-Generation Inhibitors of Bruton Tyrosine Kinase. J. Hematol. Oncol. 2016, 9, 80.
- Norman, P. Investigational Bruton’s Tyrosine Kinase Inhibitors for the Treatment of Rheumatoid Arthritis. Expert Opin. Investig. Drugs 2016, 25, 891–899.
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252.
- Lou, Y.; Owens, T.D.; Kuglstatter, A.; Kondru, R.K.; Goldstein, D.M. Bruton’s Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evaluation for Inflammatory Diseases and B Cell Malignancies. J. Med. Chem. 2012, 55, 4539–4550.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
01 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No