Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Teresa Suarez | -- | 1511 | 2022-08-30 16:08:22 | | | |
| 2 | Lindsay Dong | + 2 word(s) | 1513 | 2022-08-31 03:07:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Suarez, T.; Lindo, N.A.; Rosa, E.J.J.D.L. DNA Double Strand Breaks in Developing Neuroretina. Encyclopedia. Available online: https://encyclopedia.pub/entry/26682 (accessed on 09 August 2026).
Suarez T, Lindo NA, Rosa EJJDL. DNA Double Strand Breaks in Developing Neuroretina. Encyclopedia. Available at: https://encyclopedia.pub/entry/26682. Accessed August 09, 2026.
Suarez, Teresa, Noemi Alvarez Lindo, Enrique J J. De La Rosa. "DNA Double Strand Breaks in Developing Neuroretina" Encyclopedia, https://encyclopedia.pub/entry/26682 (accessed August 09, 2026).
Suarez, T., Lindo, N.A., & Rosa, E.J.J.D.L. (2022, August 30). DNA Double Strand Breaks in Developing Neuroretina. In Encyclopedia. https://encyclopedia.pub/entry/26682
Suarez, Teresa, et al. "DNA Double Strand Breaks in Developing Neuroretina." Encyclopedia. Web. 30 August, 2022.
Copy Citation
The existence of double strand breaks (DSBs) in the developing mouse retina is an inherent feature of the process and it is related to the early death that occurs during development. This early cell death facilitates the selection of the fittest neurons to complete the developing process successfully. In addition to the DNA repair mutants, which are clearly involved in the DSBs repair process, the fact that the rag2 endonuclease mutant also displays a similar phenotype suggests that this process may be at the basis of the somatic variability present in mammalian neurons
DSBs
neuron somatic mosaicism
Rag2
recombination
1. Introduction
In the last decade, new single cell sequencing technologies have completely changed the understanding of neural DNA, revealing that almost all healthy individuals studied carry large numbers of neuron-specific genetic alterations, most of which require prior generation and repair of DNA double strand breaks (DSBs) [1][2][3][4][5][6][7][8]. This variability is far more frequent than ever expected. Single cell genomics has demonstrated somatic mosaicism in physiological contexts in more than 10% of neurons within a given individual, increasing to 90% in some studies [1][2][3][4]. These findings indicate that cell heterogeneity in the central nervous system (CNS) relies not only on transcriptional, morphological and functional diversity, but also on major, likely underlying, changes in neuronal DNA.
Neuronal DNA is enriched as a consequence of multiple genetic alterations in neural progenitor cells. Affected regions range in size from over 500 Mb to single nucleotides (single nucleotides variations, SNVs) (Figure 1). These major genetic alterations include indels (insertion/deletion), MEIs (mobile element insertions), CNVs (copy number variations), SVs (structural variants), and aneuploidy [9]. Advances in single-cell whole-genome sequencing have provided detailed information about smaller CNVs (<1 Mb) and have shown that these are very frequent in the developing cerebral cortex [10]. All these forms of somatic mosaicism require prior generation and repair of DNA DSBs, which in turn must be properly repaired to prevent programmed cell death of the affected neuronal cell.
This somatic mosaicism within neurons results in changes in their gene expression [11], although these changes do not necessarily affect neuronal connectivity or survival in the adult brain [12]. Particularly, aneuploid neurons have been found to be active and fully integrated in the normal adult mammalian brain [12][13]. Moreover, the recent discovery of abundant CNVs of <1 Mb revealed quantitative variation at particular developmental stages in the mouse cerebral cortex [10], suggesting that these alterations may constitute part of a process intrinsic to neural development.
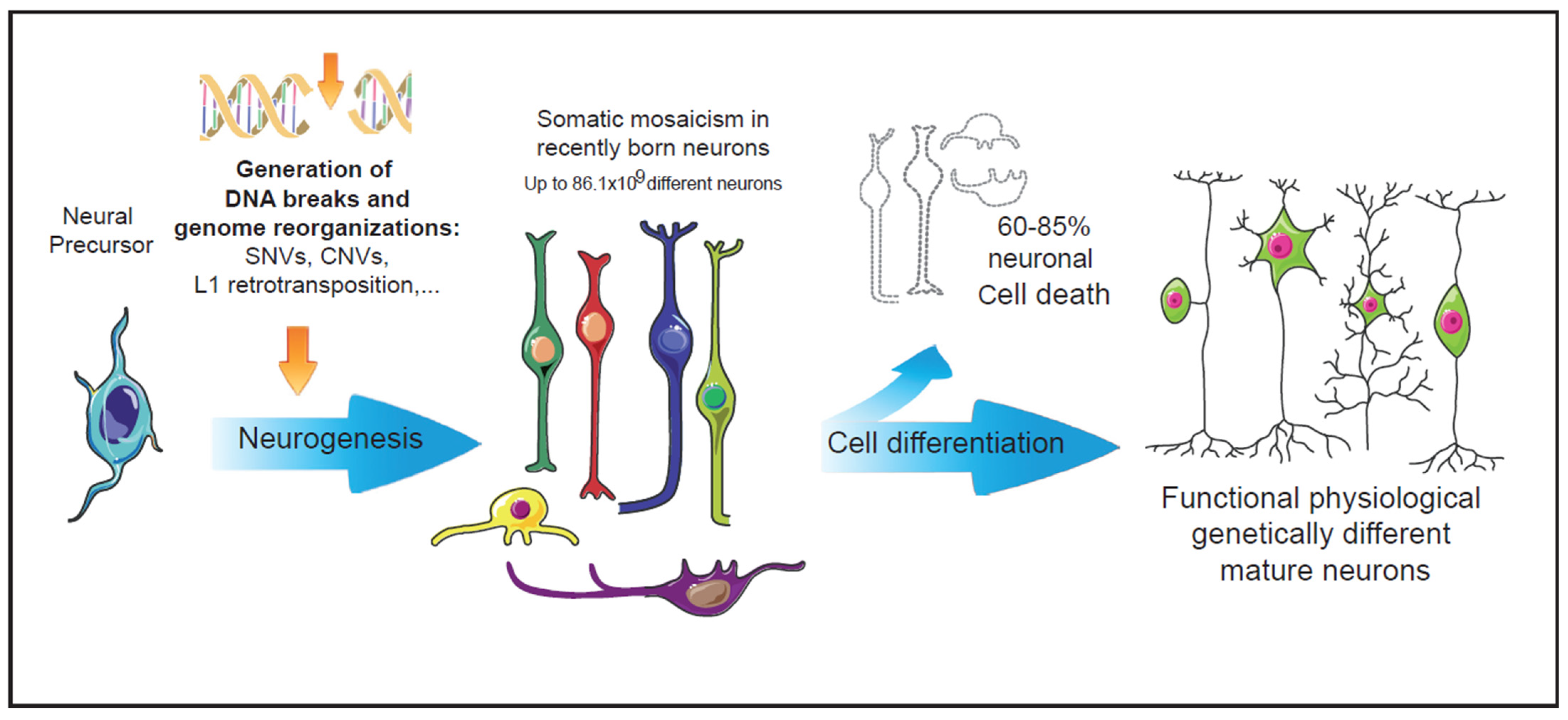
Figure 1. Intrinsic events during early neurogenesis that may contribute to somatic mosaicism and functional diversity in the mature nervous system. In the nervous system, neuronal genetic diversity seems to arise intrinsically during early neuronal differentiation. Diverse genetic alterations have been observed in healthy neurons, most of them involving the generation and repair of DNA double strand breaks. The pool of neurons with cell-unique differences in their DNA could be even larger than the vast numbers of antibodies generated by the V(d)J recombination in the immune system [14]. During neuronal differentiation, more than 60% of the recently generated neurons undergo cell death events [15]. The surviving neurons, many of them carrying genetic alterations, may configure a functional repertoire characterized by the physiologically generated somatic mosaicism.
2. DSBs and Neural Development
DSBs can result from extrinsic causes, including certain viruses, ionizing radiation, and chemical sources, or from intrinsic causes, such as reactive oxygen species produced by cellular respiration or replication fork collapse at genome fragile sites [16][17][18][19]. Alternatively, they can be a consequence of specific mechanisms and examples include programmed genome reorganization, such as RAG-1,2 endonuclease-mediated V(D)J somatic recombination, as it occurs in the immune system [20][21]. In the brain, DSBs can also be formed by specific mechanisms, such as retrotransposon mobilization (e.g., LINE-1 transposition in neural progenitors and mature neurons [22][23]) and RNA retroinsertion, as in somatic APP gene recombination [24]. Specific DSB generation mediated by endonucleases to control gene expression, as described for DNA topoisomerase II β (TOP2β) and Spo11 in the promotors of neuronal activity-induced genes [25][26][27], have also been shown.
DSBs constitute a potentially serious threat to cell survival and, therefore, must be properly repaired. DSBs are repaired by either homologous recombination or nonhomologous end-joining (NHEJ). NHEJ involves the direct ligation of the two DNA ends that frequently alter the original DNA sequence. A defective DNA damage response severely impacts nervous system development [28], and alterations in factors involved in the DSB response have been implicated in a range of diverse human syndromes, including neuropathology and neurodegeneration (e.g., ataxia telangiectasia) [29][30][31]. The importance of DSBs and their consequences for neurons are clearly evidenced by the phenotype of mice carrying DNA repair mutations. These mice present a marked, and in some cases lethal, embryonic phenotype, characterized by a high level of neuronal cell death, impaired development, and even acellularity in the central nervous system [15][32]. Indeed, mouse models with NHEJ mutations (e.g., XRCC4 and DNA lig IV) display a dramatic neural phenotype, in some cases with embryonic lethality [33][34], and severe immunodeficiency, although the remaining organs and tissues are largely unaffected, suggesting an important role of NHEJ in neural development.
Together, the neurogenic process provides an environment permissive to DSB generation and subsequent genetic alterations. The specificity of these neural DNA alterations is supported by the observation that genetic changes emerge specifically during in vitro stem cell differentiation towards neural lineage [3] but are completely absent during differentiation towards fibroblasts. This accumulation of DSBs during neural development suggests that neurons possess mechanisms to cope with DSBs, which may even have a particular function or be generated as a side-effect of another, as-yet-unknown process.
3. DSBs and Early Neural Cell Death
Several studies have shown that the number of neurons with somatic mutations decreases after development; both the number of CNVs per neuron [3] and the number of aneuploid neurons [35][36][37] are lower in the newborn and young adult brain than during embryonic development [9]. These observations suggest the involvement of cell death processes, which may selectively target non-viable mutations in a manner analogous to apoptosis following V(D)J recombination [38]. In fact, many authors, ourselves included, have shown that neural precursors and newborn neurons actually undergo a specific wave of apoptosis during early embryonic development [32][39][40][41][42]. Although the purpose of this early wave of neuronal cell death has not been clearly established, the findings in mutant models that lack apoptosis genes have underscored its importance. The dysregulation of cell death in this context specifically impairs proper CNS generation, affecting neural precursor cell proliferation and early neuronal differentiation [15][39][43]. Specifically, the dysregulation of cell death results in neural malformations including cerebral hyperplasia, exencephaly, and neural tube defects, as well as defective retinal structure and visual system connectivity [15][32][39][44][45][46].
Early neuronal cell death in the mouse retina parallels the onset of DSB generation (Figure 2), and that apoptosis occurs during a specific time window [47]. In the developing chicken retina, the onset of cell death is carefully programmed and intrinsically determined, specifically the death of newborn neurons is determined, in a cell-autonomous manner, by the time the neuron is generated and it is independent of the niche in which it was situated [48][49].
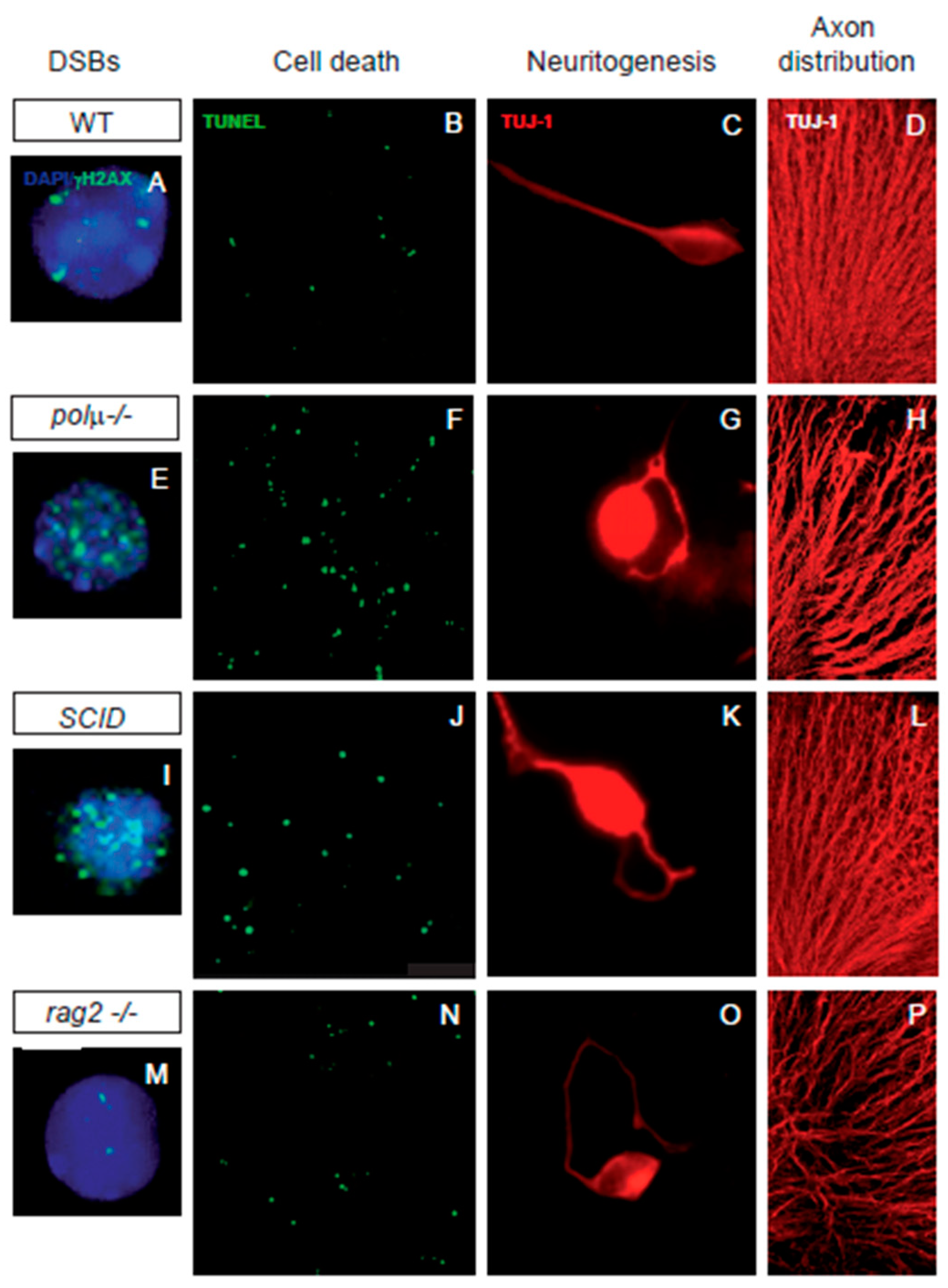
Figure 2. Impaired retinal development in mutant mice defective in components of DSB generation and repair mechanisms. Comparison of retinal phenotypes in mutant mice defective in DNA polymerase mu (polµ-/-; E-H), DNA-PK (SCID; I-L), and one of the subunits of the RAG-1,2 endonuclease responsible for generating the DSBs that originate the V(d)J recombination in the immune system (rag2-/-; M-P). The phenotype of the WT mouse is displayed in panels (A–D,A,E,I,M) E13.5 dissociated retinal cells were immunostained for γH2AX (cyan) to reveal DSBs and counterstained with DAPI (blue) to visualize the nuclei. Notice that the foci numbers are increased in the repair defective mutants and reduced in the RAG2 defective mutant, with respect to the WT. (B,F,J,N) programmed cell death was detected by TUNEL (green) in whole mount E13.5 retinas. Notice that apoptotic nuclei numbers are increased in all three mutant mice with respect to the WT mouse. (C,G,K,O) E13.5 dissociated retinal cells were cultured on polyornithine/laminin-treated plates. Neurite emission was visualized by TUJ-1 immunostaining (red). Notice the disturbed axonal trajectories in all three mutant mice, with respect to the WT mouse. (D,H,L,P) E13.5 whole-mount retinas were immunostained with TUJ-1 (red) to visualize RGC axonal trajectories. Notice the disturbed axonal trajectories in all three mutant mice, with respect to the WT mouse. Images adapted from [47][50][51][52].
In neural development, DSB repair is a key step in controlling DNA rearrangements during neurogenesis, the failure of which results in programmed cell death [30][53][54]. Several findings suggest a close association between neuronal cell death and NHEJ, the main DSB repair pathway in neurons. Mice with impaired NHEJ show a dramatic increase in programmed cell death in neural tissues, frequently resulting in embryonic or perinatal lethality. The phenotypes found in NHEJ repair mutants suggest that DSB generation and repair significantly influence the dynamics of neural development to promote neural diversity. Mice deficient in NHEJ proteins, such as KU-86, DNA polymerase mu, and DNA-PK, share a mild retinal phenotype characterized by moderate neuronal cell death ([55][56][57], Figure 2).
References
- Evrony, G.D.; Cai, X.; Lee, E.; Hills, L.B.; Elhosary, P.C.; Lehmann, H.S.; Parker, J.J.; Atabay, K.D.; Gilmore, E.C.; Poduri, A.; et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012, 151, 483–496.
- Evrony, G.D.; Lee, E.; Mehta, B.K.; Benjamini, Y.; Johnson, R.M.; Cai, X.; Yang, L.; Haseley, P.; Lehmann, H.S.; Park, P.J.; et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron 2015, 85, 49–59.
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637.
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2015, 10, 645.
- Bushman, D.M.; Chun, J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin. Cell Dev. Biol. 2013, 24, 357–369.
- Johnson, M.B.; Walsh, C.A. Cerebral cortical neuron diversity and development at single-cell resolution. Curr. Opin. Neurobiol. 2017, 42, 9–16.
- Westra, J.W.; Rivera, R.R.; Bushman, D.M.; Yung, Y.C.; Peterson, S.E.; Barral, S.; Chun, J. Neuronal DNA content variation (DCV) with regional and individual differences in the human brain. J. Comp. Neurol. 2010, 518, 3981–4000.
- Hazen, J.L.; Faust, G.G.; Rodriguez, A.R.; Ferguson, W.C.; Shumilina, S.; Clark, R.A.; Boland, M.J.; Martin, G.; Chubukov, P.; Tsunemoto, R.K.; et al. The Complete Genome Sequences, Unique Mutational Spectra, and Developmental Potency of Adult Neurons Revealed by Cloning. Neuron 2016, 89, 1223–1236.
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048.
- Rohrback, S.; April, C.; Kaper, F.; Rivera, R.R.; Liu, C.S.; Siddoway, B.; Chun, J. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2018, 115, 10804–10809.
- Kaushal, D.; Contos, J.J.A.; Treuner, K.; Yang, A.H.; Kingsbury, M.A.; Rehen, S.; McConnell, M.J.; Okabe, M.; Barlow, C.; Chun, J. Alteration of Gene Expression by Chromosome Loss in the Postnatal Mouse Brain. J. Neurosci. 2003, 23, 5599–5606.
- Kingsbury, M.A.; Friedman, B.; McConnell, M.J.; Rehen, S.K.; Yang, A.H.; Kaushal, D.; Chun, J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Natl. Acad. Sci. USA 2005, 102, 6143–6147.
- Yang, A.H.; Kaushal, D.; Rehen, S.; Kriedt, K.; Kingsbury, M.A.; McConnell, M.J.; Chun, J. Chromosome Segregation Defects Contribute to Aneuploidy in Normal Neural Progenitor Cells. J. Neurosci. 2003, 23, 10454–10462.
- Muotri, A.R.; Gage, F.H. Generation of neuronal variability and complexity. Nature 2006, 441, 1087–1093.
- Boya, P.; de la Rosa, E.J. Cell death in early neural life. Birth Defects Res. C Embryo Today 2005, 75, 281–293.
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246.
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211.
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510.
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455.
- Lescale, C.; Deriano, L. The RAG recombinase: Beyond breaking. Mech. Ageing Dev. 2017, 165, 3–9.
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429.
- Thomas, C.A.; Paquola, A.C.; Muotri, A.R. LINE-1 retrotransposition in the nervous system. Annu. Rev. Cell Dev. Biol. 2012, 28, 555–573.
- Suarez, N.A.; Macia, A.; Muotri, A.R. LINE-1 retrotransposons in healthy and diseased human brain. Dev. Neurobiol. 2017, 78, 434–455.
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645.
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802.
- Puc, J.; Aggarwal, A.K.; Rosenfeld, M.G. Physiological functions of programmed DNA breaks in signal-induced transcription. Nat. Rev. Mol. Cell Biol. 2017, 18, 471–476.
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613.
- Frappart, P.O.; McKinnon, P.J. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair 2008, 7, 1051–1060.
- McKinnon, P.J.; Caldecott, K.W. DNA strand break repair and human genetic disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 37–55.
- Lee, Y.; McKinnon, P.J. Responding to DNA double strand breaks in the nervous system. Neuroscience 2007, 145, 1365–1374.
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529.
- Valenciano, A.I.; Boya, P.; de la Rosa, E.J. Early neural cell death: Numbers and cues from the developing neuroretina. Int. J. Dev. Biol. 2009, 53, 1515–1528.
- Gao, Y.; Sun, Y.; Frank, K.M.; Dikkes, P.; Fujiwara, Y.; Seidl, K.J.; Sekiguchi, J.M.; Rathbun, G.A.; Swat, W.; Wang, J.; et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 1998, 95, 891–902.
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell 2000, 5, 993–1002.
- Rehen, S.K.; McConnell, M.J.; Kaushal, D.; Kingsbury, M.A.; Yang, A.H.; Chun, J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. USA 2001, 98, 13361–13366.
- McConnell, M.J.; Kaushal, D.; Yang, A.H.; Kingsbury, M.A.; Rehen, S.K.; Treuner, K.; Helton, R.; Annas, E.G.; Chun, J.; Barlow, C. Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J. Neurosci. 2004, 24, 8090–8096.
- Rehen, S.K.; Yung, Y.C.; McCreight, M.P.; Kaushal, D.; Yang, A.H.; Almeida, B.S.; Kingsbury, M.A.; Cabral, K.M.; McConnell, M.J.; Anliker, B.; et al. Constitutional aneuploidy in the normal human brain. J. Neurosci. 2005, 25, 2176–2180.
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200.
- Yeo, W.; Gautier, J. Early neural cell death: Dying to become neurons. Dev. Biol. 2004, 274, 233–244.
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174.
- de la Rosa, E.J.; de Pablo, F. Cell death in early neural development: Beyond the neurotrophic theory. Trends Neurosci. 2000, 23, 454–458.
- Chavarria, T.; Baleriola, J.; Mayordomo, R.; de Pablo, F.; de la Rosa, E.J. Early neural cell death is an extensive, dynamic process in the embryonic chick and mouse retina. Sci. World J. 2013, 2013, 627240.
- Buss, R.R.; Sun, W.; Oppenheim, R.W. Adaptive roles of programmed cell death during nervous system development. Annu. Rev. Neurosci. 2006, 29, 1–35.
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809.
- Kaufmann, S.H.; Hengartner, M.O. Programmed cell death: Alive and well in the new millennium. Trends Cell Biol. 2001, 11, 526–534.
- Mayordomo, R.; Valenciano, A.I.; de la Rosa, E.J.; Hallbook, F. Generation of retinal ganglion cells is modulated by caspase-dependent programmed cell death. Eur. J. Neurosci. 2003, 18, 1744–1750.
- Baleriola, J.; Suarez, T.; de la Rosa, E.J. DNA-PK promotes the survival of young neurons in the embryonic mouse retina. Cell Death Differ. 2010, 17, 1697–1706.
- Galli-Resta, L.; Ensini, M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J. Neurosci. 1996, 16, 2318–2324.
- Southwell, D.G.; Paredes, M.F.; Galvao, R.P.; Jones, D.L.; Froemke, R.C.; Sebe, J.Y.; Alfaro-Cervello, C.; Tang, Y.; Garcia-Verdugo, J.M.; Rubenstein, J.L.; et al. Intrinsically determined cell death of developing cortical interneurons. Nature 2012, 491, 109–113.
- Alvarez-Lindo, N.; Baleriola, J.; de Los Rios, V.; Suarez, T.; de la Rosa, E.J. RAG-2 deficiency results in fewer phosphorylated histone H2AX foci, but increased retinal ganglion cell death and altered axonal growth. Sci. Rep. 2019, 9, 18486.
- Baleriola, J.; Alvarez-Lindo, N.; de la Villa, P.; Bernad, A.; Blanco, L.; Suarez, T.; de la Rosa, E.J. Increased neuronal death and disturbed axonal growth in the Polmu-deficient mouse embryonic retina. Sci. Rep. 2016, 6, 25928.
- Alvarez-Lindo, N. Implicación de la Generación y Reparación de Roturas del DNA en el Desarrollo de la Retina de Ratón. Ph.D. Thesis, Universidad Complutense de Madrid, Madrid, Spain, 2015.
- Chun, J.J.; Schatz, D.G.; Oettinger, M.A.; Jaenisch, R.; Baltimore, D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell 1991, 64, 189–200.
- Gilmore, E.C.; Nowakowski, R.S.; Caviness, V.S., Jr.; Herrup, K. Cell birth, cell death, cell diversity and DNA breaks: How do they all fit together? Trends Neurosci. 2000, 23, 100–105.
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair 2014, 16, 84–96.
- Zha, S.; Alt, F.W.; Cheng, H.L.; Brush, J.W.; Li, G. Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4518–4523.
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171.
More
Information
Subjects:
Developmental Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
706
Revisions:
2 times
(View History)
Update Date:
06 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No