Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Firoozeh Shomal Zadeh | -- | 3218 | 2022-08-29 18:58:30 | | | |
| 2 | Catherine Yang | Meta information modification | 3218 | 2022-08-30 03:18:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shafiei, M.; Zadeh, F.S.; Mansoori, B.; Pyle, H.; Agim, N.; Hinojosa, J.; Dominguez, A.; Thomas, C.; Chalian, M. Imaging More than Skin-Deep. Encyclopedia. Available online: https://encyclopedia.pub/entry/26636 (accessed on 28 July 2026).
Shafiei M, Zadeh FS, Mansoori B, Pyle H, Agim N, Hinojosa J, et al. Imaging More than Skin-Deep. Encyclopedia. Available at: https://encyclopedia.pub/entry/26636. Accessed July 28, 2026.
Shafiei, Mehrzad, Firoozeh Shomal Zadeh, Bahar Mansoori, Hunter Pyle, Nnenna Agim, Jorge Hinojosa, Arturo Dominguez, Cristina Thomas, Majid Chalian. "Imaging More than Skin-Deep" Encyclopedia, https://encyclopedia.pub/entry/26636 (accessed July 28, 2026).
Shafiei, M., Zadeh, F.S., Mansoori, B., Pyle, H., Agim, N., Hinojosa, J., Dominguez, A., Thomas, C., & Chalian, M. (2022, August 29). Imaging More than Skin-Deep. In Encyclopedia. https://encyclopedia.pub/entry/26636
Shafiei, Mehrzad, et al. "Imaging More than Skin-Deep." Encyclopedia. Web. 29 August, 2022.
Copy Citation
Systemic disorders often present with both cutaneous and radiologic findings. Some presenting skin lesions are disease-specific (e.g., shagreen patch in tuberous sclerosis); others signify internal organ involvement (e.g., erythema nodosum in sarcoidosis, tuberculosis, or inflammatory bowel disease). Disease entities are broadly categorized into three areas: (1) autoimmune/inflammatory disorders and vasculitides, (2) genetic/congenital disorders, and (3) neoplasms.
dermatology
systemic
cutaneous
radiologic features
1. Dermatomyositis
Dermatomyositis (DM) is a rare autoimmune disease occurring with a bimodal peak incidence of 5–15 at 40–60 years of age with a 2:1 female: male predominance. DM is characterized by skin lesions and myositis [1][2][3]. Skin lesions in DM are often pruritic or burning, photosensitive, and precede myopathic symptoms in 50% of patients [2][4]. Atrophic dermal papules of dermatomyositis (ADPDM, formerly Gottron papules) are pathognomonic violaceous papules and plaques, sometimes with subtle scale, found on the interphalangeal joints of the hands (Figure 1A) [4]. Additional pathognomonic findings include Gottron sign (erythematous macules or patches over the extensor joints) and the heliotrope rash (periorbital erythema with edema, most often affecting the upper eyelids) [4]. Other characteristic skin findings in DM include V sign (erythematous, confluent papules, and plaques over the lower anterior neck and upper chest), shawl sign (violaceous or erythematous papules and plaques over the posterior shoulders and upper back), calcinosis cutis, and nailfold changes (periungual erythema, capillary loop dilation and dropout, and ragged cuticles) [2][3][4].
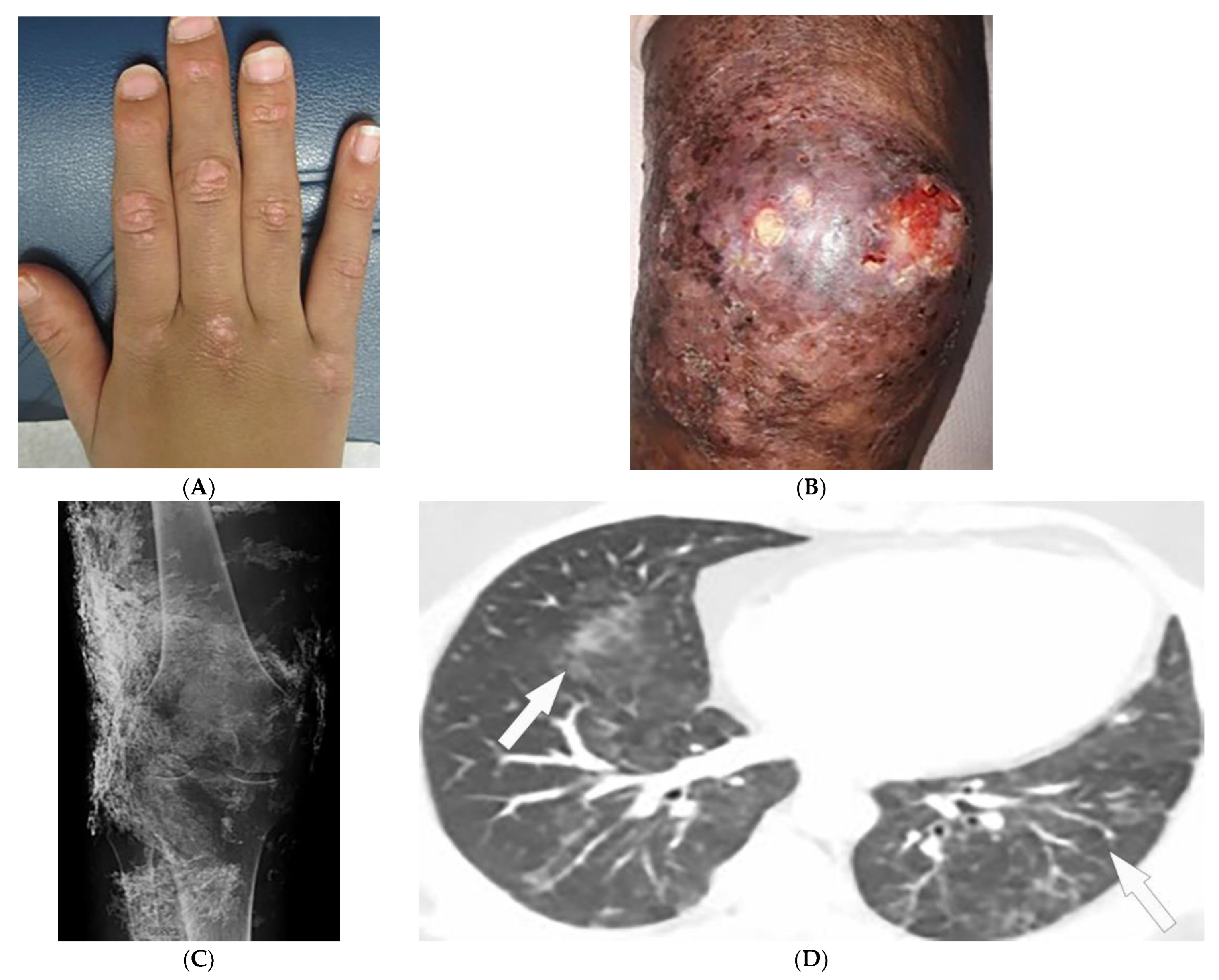
Figure 1. Dermatologic and radiologic images representative of dermatomyositis: (A) Flat-topped papules “Gottron’s papules” over the dorsum of the hand of a 19-year-old female. (B) Skin erythema and ulceration of the knee due to soft-tissue calcifications in a 45-year-old female. (C) AP radiograph of the same knee displays sheet-like soft-tissue calcifications. (D) Axial chest CT image (lung window) demonstrates patchy bilateral ground-glass opacities (GGOs) indicative of dermatomyositis-associated interstitial lung disease (ILD) (arrows).
A large body of evidence exists substantiating an association between dermatomyositis and malignancy, and newly diagnosed patients should be screened for underlying malignancy. Screening typically involves a comprehensive review of the patient’s history, physical examination, and basic labs followed by CT imaging of chest/abdomen/pelvis in cases of high suspicion. Features associated with an increased risk of malignancy include older age at onset (>45), male sex, dysphagia, cutaneous necrosis, cutaneous vasculitis, rapid onset myositis, and elevated inflammatory markers [5][6].
Magnetic resonance imaging (MRI) has become a fundamental noninvasive tool for assessment of myositis. MRI provides information to diagnose subclinical disease, estimate disease chronicity, detect optimal site for biopsy, and evaluates treatment response. However, its application is still limited due to cost and availability [1][6][7][8]. Multifocal areas of hyperintensities on fluid-sensitive sequences and areas of enhancement on contrast-enhanced fat-saturated T1-weighted images (T1WI) are seen in active disease. Fatty atrophy (chronic disease) is best visualized on T1-weighted sequences [6][7][8]. Soft-tissue calcifications with varying patterns, including nodular, reticular, amorphous, and sheet-like, occur in up to 70% of patients with DM, predominantly children (Figure 1B). If there is diagnostic uncertainty, plain radiographs are recommended for the initial detection of soft-tissue calcinosis due to their high sensitivity, availability, and low cost (Figure 1C) [1].
Thoracic complications occur in >50% of patients and include interstitial lung disease (ILD), aspiration pneumonia, and hypoventilation [3]. While ILD can present as reticulonodular opacities on radiograph, high-resolution computed tomography (HRCT) of the chest is able to differentiate between predominant patterns with higher diagnostic accuracy. These patterns of ILD, which may coexist (Figure 1D) [1] include (A) nonspecific interstitial pneumonia (NSIP, with imaging characteristics of ground-glass opacification (GGO) with reticulation and traction bronchiectasis), (B) organizing pneumonia (OP, seen as peripheral bilateral consolidation and patchy GGOs) and to a lesser extent (C) usual interstitial pneumonia (UIP, manifesting as peripheral reticulation, traction bronchiectasis, and honeycombing), all with basilar predilection. Another complication of DM is the involvement of pharyngeal muscles, which may result in dysphagia, and aspiration pneumonia. This could be measured by an esophagogram, which is a dynamic fluoroscopic swallow study [1][3].
2. Sarcoidosis
Sarcoidosis is a granulomatous disease with unknown etiology and variable prevalence, with a predilection for African American women in their third to fifth decades of life [9][10][11][12]. Patients may be asymptomatic or experience a wide spectrum of multiorgan involvement [10][11] demanding radiologic investigation for diagnosis and follow-up [9][10][12]. Twenty to thirty-five percent of patients with sarcoidosis develop skin lesions, which often manifest at the onset of systemic illness and, thus, can provide diagnostic clues. Specific skin lesions are those with epithelioid granulomas without associated inflammation on histopathology (e.g., lupus pernio, Darier–Roussy). Erythema nodosum (EN, reported in up to 25% of cases) consists of tender self-limiting erythematous subcutaneous nodules most commonly present on the shin. Lupus pernio (LP) describes characteristic chronic violaceous indurated papulonodules with central face distribution. It is strongly associated with extracutaneous involvement, specifically pulmonary disease [9][11][12][13]. Sarcoid may also present as firm, well-demarcated, skin-colored to violaceous papules with a predilection for the face (Figure 2A). Of note, sarcoidosis has been reported to mimic other diseases, including herpes zoster, chronic cutaneous lupus erythematosus, ichthyosis, and psoriasis [14].
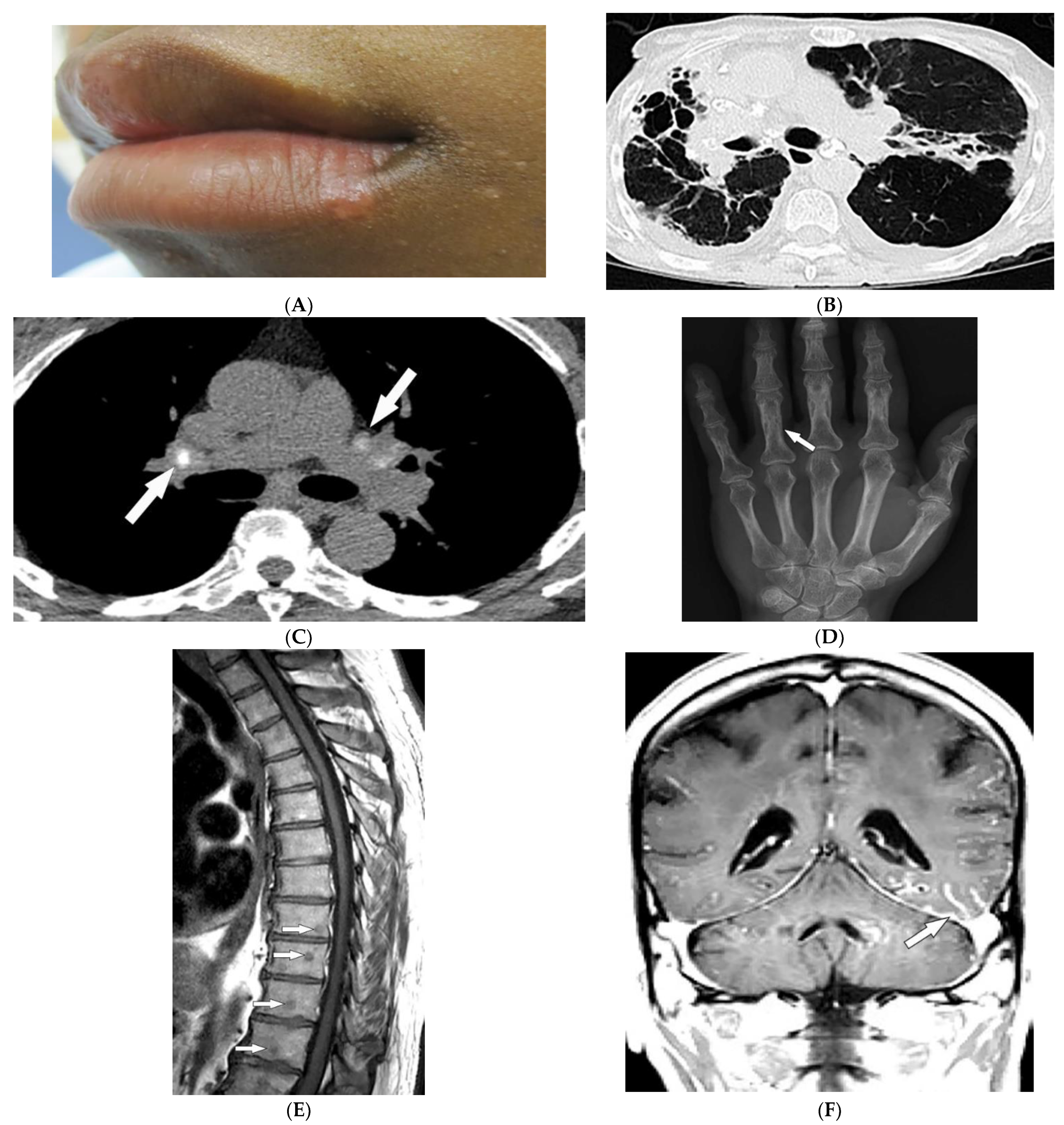
Figure 2. Dermatologic and radiologic images characteristic of sarcoidosis: (A) Skin-colored papules on mucosal and cutaneous lips seen in a 19-year-old male with skin sarcoidosis. (B) Axial chest HRCT image (lung window) of a 56-year-old male demonstrates multiple areas of bronchiectasis, cysts, and architectural distortion consistent with end-stage pulmonary sarcoidosis. (C) Axial chest CT image (soft-tissue window) in a 35-year-old male showing calcified hilar lymph nodes (arrows). (D) AP radiograph of the hand of a 45-year-old male demonstrates lacy lytic osseous sarcoid of multiple phalanges (arrow). (E) Sagittal T1-weighted image (T1WI) of the thoracic spine in a 40-year-old woman shows multiple well-circumscribed sarcoid marrow lesions (arrows). (F) Coronal contrast-enhanced T1WI of the brain of a 42-year-old male shows leptomeningeal enhancement (arrow).
Thoracic involvement is seen in nearly 90% of patients with chronic sarcoidosis, and usually presents with symmetric, bilateral, hilar, and right paratracheal adenopathy with amorphous or cloud-like calcified lymph nodes (Figure 2B). Parenchymal involvement and pulmonary embolism are other chest manifestations of sarcoidosis. Parenchymal involvement usually reveals bilateral, symmetric, small, rounded opacities with apical predominance on chest radiography. Irregular (2–5 mm) nodules with perilymphatic predilection causing irregular micronodular thickening of fissures and interlobular septa can be seen on HRCT. Parenchymal involvement can progress to irreversible disease manifesting as mid- to upper lung reticular opacities radiating from the hila (Figure 2C) [10][11][15][16][17].
Bone involvement mostly affects phalanges and toes with a pathognomonic lacy lytic appearance (Figure 2D), or less commonly, purely lytic lesions. MRI is more sensitive when evaluating appendicular skeleton, axial skeleton, and marrow involvement. MRI can show variable-sized with T1 hypointensities and T2 hyperintensities (Figure 2E). Chronic or healed lesions may present with signal intensities consistent with fat or fibrosis [10][11][12][15]. Cardiac sarcoidosis accounts for up to 85% of sarcoidosis-related deaths in Japan. Cardiac involvement patterns on late-gadolinium-enhanced cardiac MRI is nonspecific. However, it is mostly seen as patchy and multifocal late gadolinium enhancement in the basal segments of the septum and the lateral wall. On Fluorodeoxyglucose–positron emission tomography (FDG-PET), active inflammation is seen as 18F-FDG uptake with or without a perfusion defect [11].
Neurosarcoidosis (NS) can occur in any part of the brain. Cranial neuropathy is the most common CNS presentation, and the optic nerve is the most commonly involved nerve, which may be seen on MRI as thickening of the nerve with abnormal enhancement [10][11][15]. Intraparenchymal findings most commonly present as multiple small T2-hyperintense and T1-hypointense foci within the periventricular white matter. Plaque-like or nodular thickening of the hypothalamus and pituitary gland with T1 isointensity, T2 hypointensity, and marked enhancement may also present on MRI. Leptomeningeal and dural involvement are seen, respectively, as enhancing nodular (Figure 2F) and plaque-like thickening on postcontrast T1-weighted fat-saturated sequences. Dural nodularities also manifest as foci of hypointensity on T2-weighted images (T2WI). In early disease, spinal cord involvement presents as nodular leptomeningeal enhancement along the spinal cord. Late manifestations of cord involvement include elongated eccentric intramedullary T1 hypointensity and T2 hyperintensity with patchy post-contrast enhancement, typically in the cervicothoracic spine, with subsequent cord atrophy [10][11][18].
3. Scleroderma
Scleroderma, or systemic sclerosis, is an autoimmune fibrosing disorder, consisting of two subsets: diffuse cutaneous systemic sclerosis (dSSc; a systemic disease characterized by widespread involvement of any organ system with a prevalence of 20/100,000 and peak incidence in females between 30 and 50 years old), and limited cutaneous systemic sclerosis (lcSSc; a disease characterized by manifestations of the CREST syndrome (calcinosis cutis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly (Figure 3A), and telangiectasia). Cases without skin changes but other systemic manifestations have been reported as well and are described as systemic sclerosis sine scleroderma [19][20][21]. The skin is the main organ involved in scleroderma, and disease subsets are differentiated by the degree of skin involvement [20]. Raynaud’s phenomenon, cutaneous sclerosis, nailfold and fingernail alterations, cutaneous ulcerations, telangiectasias, “salt and pepper” hyper/hypopigmentation (Figure 3B), and calcinosis cutis are common skin manifestations seen in scleroderma patients [1][20]. Cutaneous sclerosis begins in the fingers, extends proximally to the metacarpophalangeal joints, and affects the face at an early stage. The skin becomes pale and hairless as the skin folds disappear. The current literature supports using high-frequency ultrasound for quantitative and reliable evaluation of dermal thickness in patients with SSc. Dermal thickness has been shown to be inversely corelated with blood perfusion. Additionally, US elastography has been shown to be of value in the evaluation of the skin in SSc [20][22][23][24][25][26]. Claw hands (Figure 3A), thin lips, sharp nose, and a characteristic “mouse face” appearance can result from this [20].
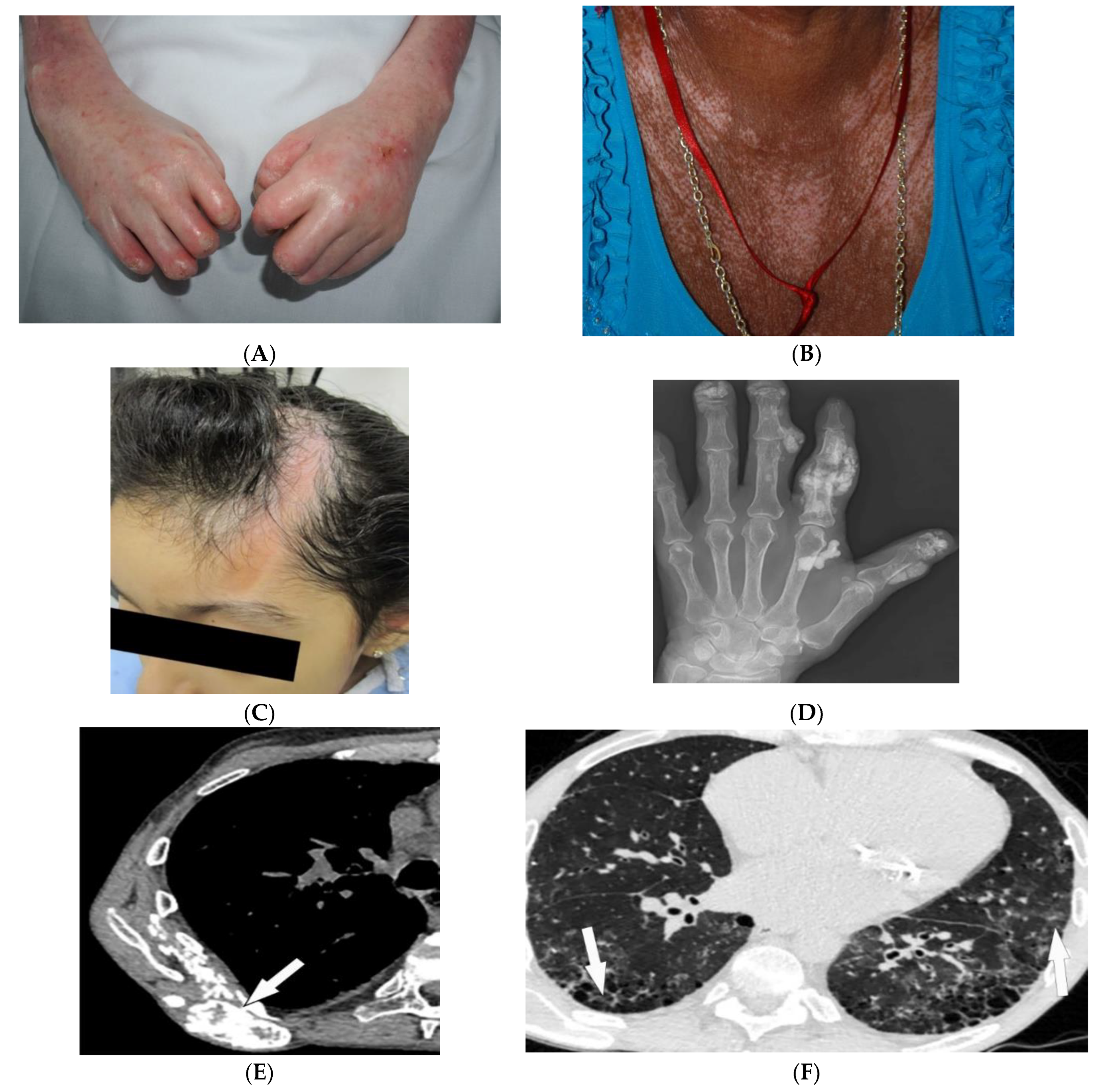
Figure 3. Dermatologic and radiologic images representative of scleroderma: (A) Taut shiny skin affecting the arms and hands with associated contractures of the fingers of a 56-year-old female patient with SSc. (B) “Salt and pepper” hyper/hypopigmentation of SSc in a 50-year-old female on the chest. (C) Indurated bound-down atrophic linear plaque with alopecia on the forehead and scalp of a 19-year-old female consistent with linear morphea. (D) AP radiograph of the hand of a 42-year-old female demonstrates soft-tissue calcifications and acro-osteolysis of the scleroderma. (E) Axial chest HRCT image (soft-tissue window) in a 42-year-old male shows the right back musculature calcinosis (arrow). (F) Axial chest HRCT image (lung window) in 38-year-old male demonstrates fine reticulonodular opacities (arrow) consistent with scleroderma-associated nonspecific interstitial pneumonia (NSIP).
Morphea, previously called localized cutaneous sclerosis (LSc), is a distinct disorder from systemic sclerosis and causes limited sclerosis of the skin (Figure 3C) with rare subcutaneous tissue and bony involvement. Patients with morphea do not typically have diffuse skin involvement or systemic manifestations. Due to confusion with SSc and to decrease patient anxiety, use of “localized scleroderma” is discouraged [19].
Soft-tissue and musculoskeletal manifestations include hand edema, acro-osteolysis (involving palmar surface with progression to pencil-tip appearance), calcinosis (Figure 3D,E), flexion contractures, and arthralgias [1][20]. Bone resorption may also be seen at the ribs, distal radius and ulna, distal clavicle, and mandible [20]. The GI tract can be involved from the esophagus to anus in patients with SSc. Up to 90% of SSc patients have significant esophageal motility abnormalities. Esophagogram shows a patulous and dilated esophagus, with no peristalsis, and the presence of gastroesophageal reflux [1][20]. Esophageal dilation and shortening below the level of the aortic arch can be seen on the classic chest radiograph. Dilated tubular esophagus without peristalsis with gastro-esophageal junction widening and contrast medium regurgitation back to the esophagus may be seen on barium swallow [26].
Pulmonary involvement is the leading cause of death in SSc and most commonly includes SSc-related ILD (SSc-ILD) and pulmonary hypertension. SSc-ILD is characterized by conventional radiography showing faint bibasilar reticulation to thick peripheral interstitial opacification accompanied by traction bronchiectasis and volume loss [20]. HRCT has a higher sensitivity for detecting SSc-ILD, including mostly NSIP and UIP [3][20]. Reticulation with a predilection for posterior basilar aspects of the lower lobes is seen in both UIP and NSIP (Figure 3F). GGOs and microcytic honeycombing are characteristic of NSIP and UIP, respectively; however, they can be found in both [1][3][20].
4. Celiac Disease
Celiac disease (CD) is an autoimmune disorder reaching a 1% incidence in most populations caused by gluten sensitivity. It is associated with intestinal and extraintestinal manifestations, including osteoporosis, iron deficiency, and skin lesions [27][28]. The most commonly associated skin manifestations are dermatitis herpetiformis (DH) and psoriasis. DH, seen in more than 85% of patients, is a chronic relapsing vesiculobullous skin disease. It is characterized by symmetric, pruritic, erythematous papules, and vesicles with a predilection for the extensor surfaces of the extremities, scalp, and buttocks (Figure 4A). Psoriasis is also associated with CD and is characterized by well-demarcated erythematous, silver-scaled plaques on extensor surfaces [27].
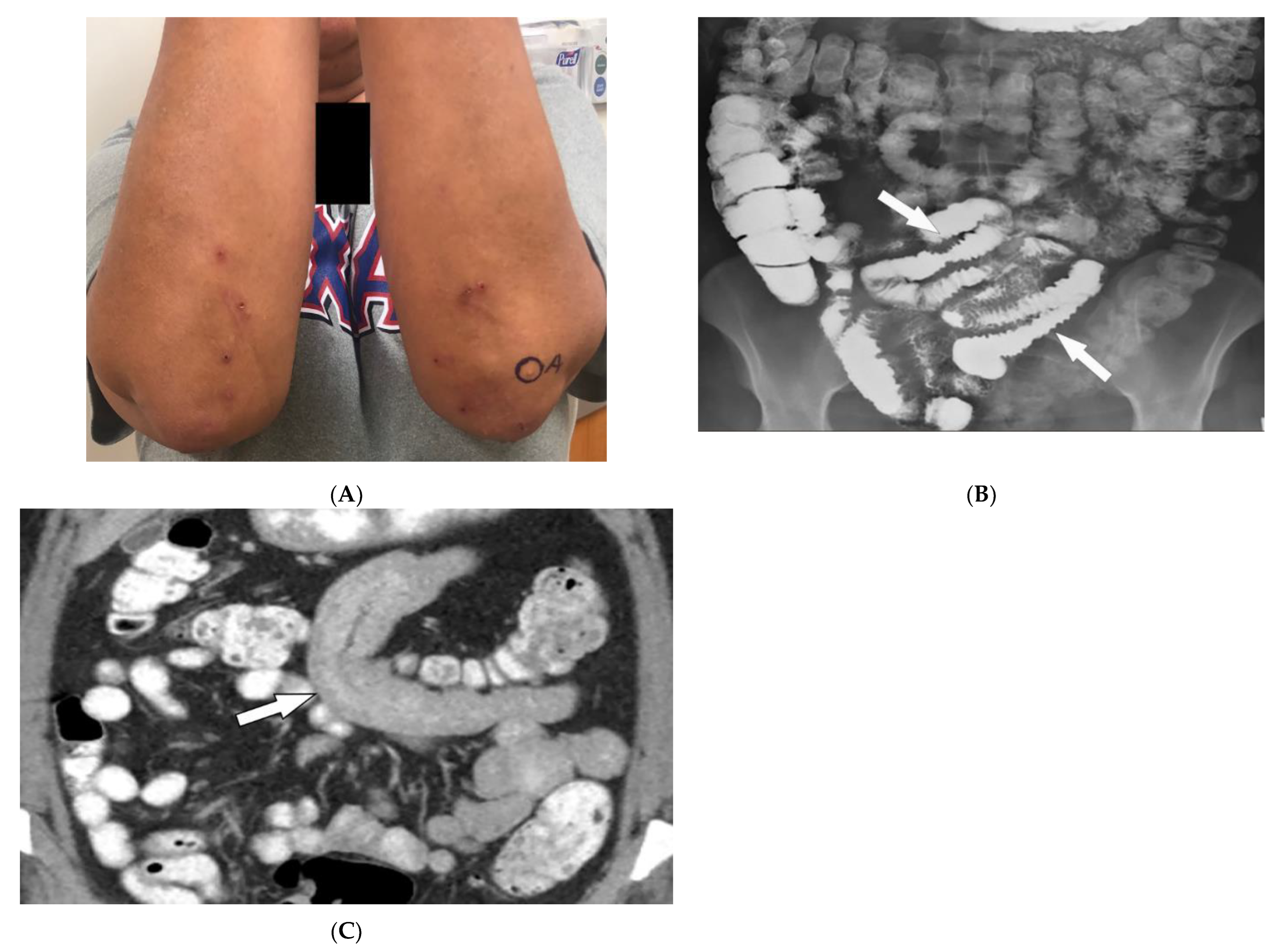
Figure 4. Dermatologic and radiologic images illustrative of Celiac disease: (A) Clustered vesicles over the bilateral extensor elbows of a 25-year-old female compatible with dermatitis herpetiformis. (B) Fluoroscopic small-bowel follow-through in a 22-year-old female demonstrates reversal of jejunal and ileal folds with more prominent folds in the ileum (arrow). (C) CT enterography coronal image (soft-tissue window) in a 42-year-old female shows thickening of the small-bowel folds (arrow).
Intestinal involvement is the leading cause of morbidity, mainly secondary to malabsorption as well as an increased risk for mucosa-associated lymphoid tissue (MALT) lymphoma [22]. Small bowel follow-through under fluoroscopy shows jejunal dilation, fold thickening, decreased jejunal fold with increased ileal fold (so-called “reversal of fold pattern”), hypomotility, and transient intussusceptions (Figure 4B) [29][30]. CT and MRI are more sensitive and can better delineate bowel wall thickening, mesenteric lymphadenopathy, duodenojejunal fatty proliferation, hypervascular mesentery, and hyposplenism. CT enterography (CTE) with intravenous and oral contrast can show ulcers, strictures, mucosal enhancement, increased splanchnic circulation, dilated vasa recta, reversed jejunoileal fold pattern, and ileal fold thickening (Figure 4C) [29][30][31]. MR enterography (MRE) has shown comparable sensitivity to CTE for the diagnosis of intestinal inflammation and has been used for the diagnosis of CD and its complications, particularly malignancy [28][30].
5. Granulomatosis with Polyangiitis
Granulomatosis with polyangiitis (GPA, formerly Wegener’s granulomatosis) is a rare necrotizing granulomatous vasculitis, which is associated with positive cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCAs). GPA primarily affects small vessels in the upper and lower respiratory tracts and kidneys, and medium-sized arteries. The peak incidence is from 46 to 60 years of age, with equal prevalence in males and females [31][32][33]. Skin involvement is seen in about 50% of patients and is polymorphous, including palpable purpura (cutaneous small vessel vasculitis is most common) (Figure 5A), nodules, vesicles, and necrotic lower extremity ulcers on a background of livedo reticularis. Pyoderma-gangrenosum-like ulcerations are a less common finding (Figure 5B) [31].
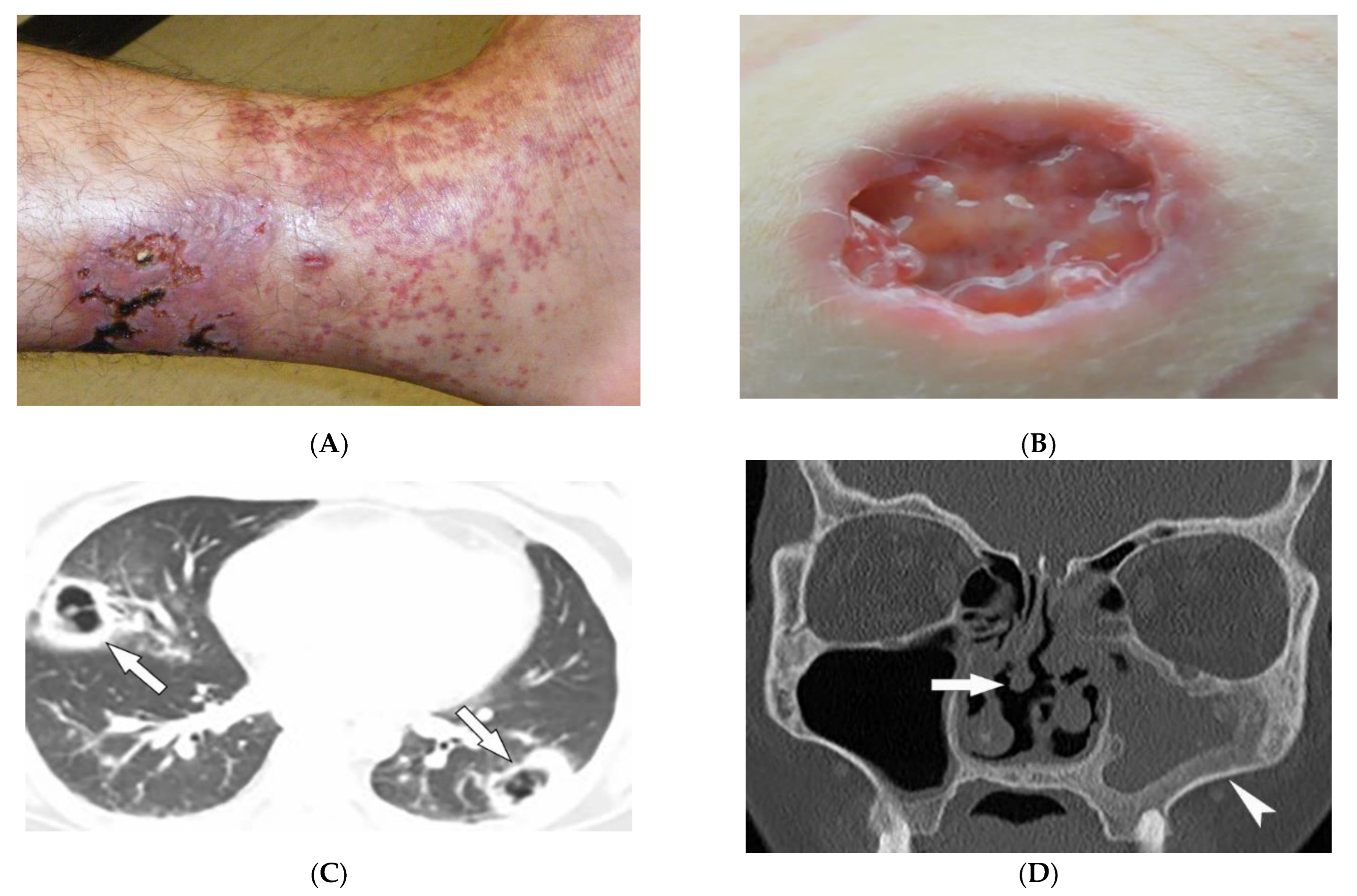
Figure 5. Dermatologic and radiologic images representative of polyangiitis: (A) Palpable cutaneous purpura with retiform eschars and ulceration in a 42-year-old male. (B) Ulcer with jagged undermined borders in a 26-year-old female with granulomatosis with polyangiitis resembling pyoderma gangrenosum. (C) Axial chest HRCT image (lung window) of a 36-year-old male demonstrates bilateral cavitary lung lesions with a ground-glass halo, suggesting surrounding hemorrhage (arrows). (D) Coronal maxillofacial CT (bone window) of a 41-year-old female shows sequelae of chronic sinusitis secondary to granulomatous inflammation, including septal perforation (arrow) and left maxillary sinus hyperostosis (arrowheads).
GPA affects the lungs in about 50–90% of patients [32]. Chest radiograph is able to detect large pulmonary nodules. HRCT shows more detailed pathologies, including variable-sized nodules (±cavitation) and GGOs (Figure 5C). Smooth or nodular thickening of the tracheobronchial tree, occasionally multifocal, causes luminal narrowing. The tracheal posterior membrane and subglottic region are most commonly involved [32][33]. Nearly all patients with GPA have ear, nose, and throat involvement at early stages of the disease [32]. Sinonasal involvement manifests as mucosal thickening, bony erosions, and neo-osteogenesis, which together are specific for GPA. Mucosal nodular thickening most commonly involves the maxillary sinuses and is mostly detected on MRI. Erosion and punctuate areas of bone destruction primarily involve the anterior ethmoidal region and is best visualized by CT [32][33]. Saddle nose deformity and perforation of the nasal septum can be present (Figure 5D) [26]. In 6% of patients, contiguous spread of inflammation can lead to the involvement of the skull base, resulting in cranial neuropathy, and is seen as cranial nerve enhancement and thickening on MRI [32][33].
6. Polyarteritis Nodosa
Polyarteritis nodosa (PAN) is a rare systemic ANCA-negative vasculitis involving small-to-medium-sized muscular-walled arteries with peak incidence in the fifth to sixth decades of life. The kidneys, skin, peripheral nerves, and gastrointestinal tract are most involved. Tissue biopsy along with clinical and laboratory data is diagnostic for PAN [33][34][35][36]. In one-third of patients, cutaneous manifestations are the primary feature of the disease. Common skin manifestations include palpable purpura, livedo reticularis, and nodules (Figure 6A). In addition, some patients might only manifest cutaneous lesions without systemic involvement, termed cutaneous PAN. The most frequent of these manifestations is the presence of nodules on the lower legs, which are often found in different stages of development. Less common skin features include urticaria, superficial phlebitis, distal necrosis, and splinter hemorrhages [36].
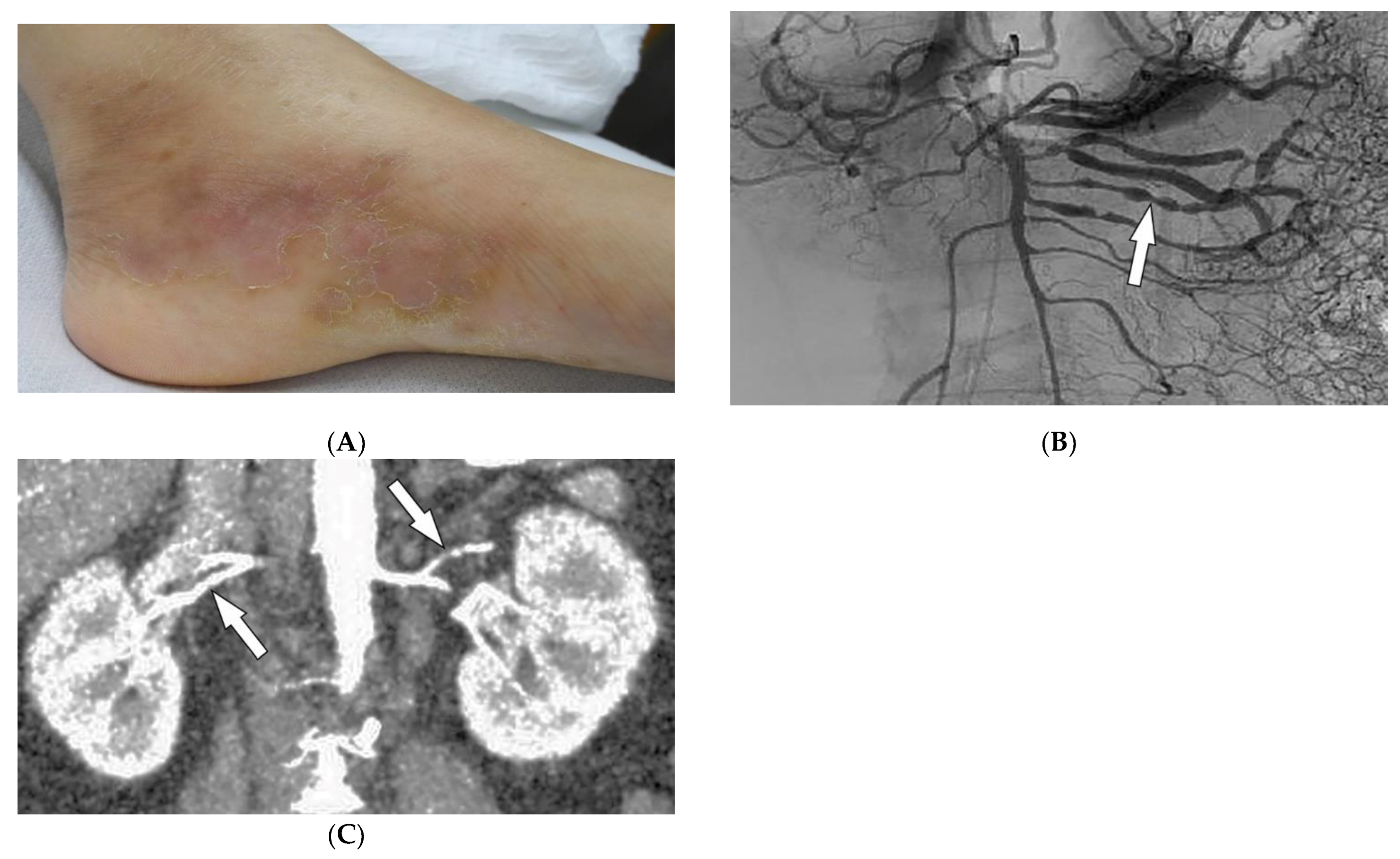
Figure 6. Dermatologic and radiologic images representative of polyarteritis nodosa: (A) Painful clustered subcutaneous nodules and plaques on the foot of a 36-year-old female. (B) Mesenteric angiogram shows the beaded appearance of multiple mesenteric arteries (arrow). (C) Coronal CT angiogram of the abdomen showed a beaded appearance of bilateral renal arteries (arrows).
Imaging can assist with the early diagnosis of PAN. Catheter angiography, CT angiography (CTA), and MR angiography (MRA) can be used to evaluate disease burden, evaluate cases where tissue biopsy is inconclusive or limited, and assess mesenteric (Figure 6B) or renal circulation (Figure 6C). Possible findings include multiple aneurysms (1–5 mm) and irregular constrictions occurring at small- and medium-sized arterial bifurcations. Unlike conventional angiography, CTA and MRA are less invasive and capable of evaluating arterial wall thickening and end-organ damage [33][34][35].
7. Behcet’s Disease
Behcet’s Disease (BD) is a systemic vasculitis of unknown etiology involving different-sized vessels presenting between the second and fourth decades of life with a higher prevalence around the historical Silk Road. Since various organs may be involved, use of appropriate imaging modalities is mandatory for the assessment of disease extent [37][38]. BD is marked by recurring oropharyngeal ulcers, genital ulcers, and ocular involvement. Earlier onset of mucocutaneous manifestations in BD indicates a worse prognosis [39]. Oral and genital involvement manifests as recurrent painful vesiculopustules evolving into apthous ulcers (Figure 7A,B). Oral ulcerations are often large and appear in groups, with frequent recurrence. Genital ulcers are smaller and occur less frequently. Other common cutaneous lesions include erythema-nodosum-like nodules, pseudofolliculitis, papulopustular lesions, acneiform nodules, and superficial thrombophlebitis (Figure 7C,D) [38][39].
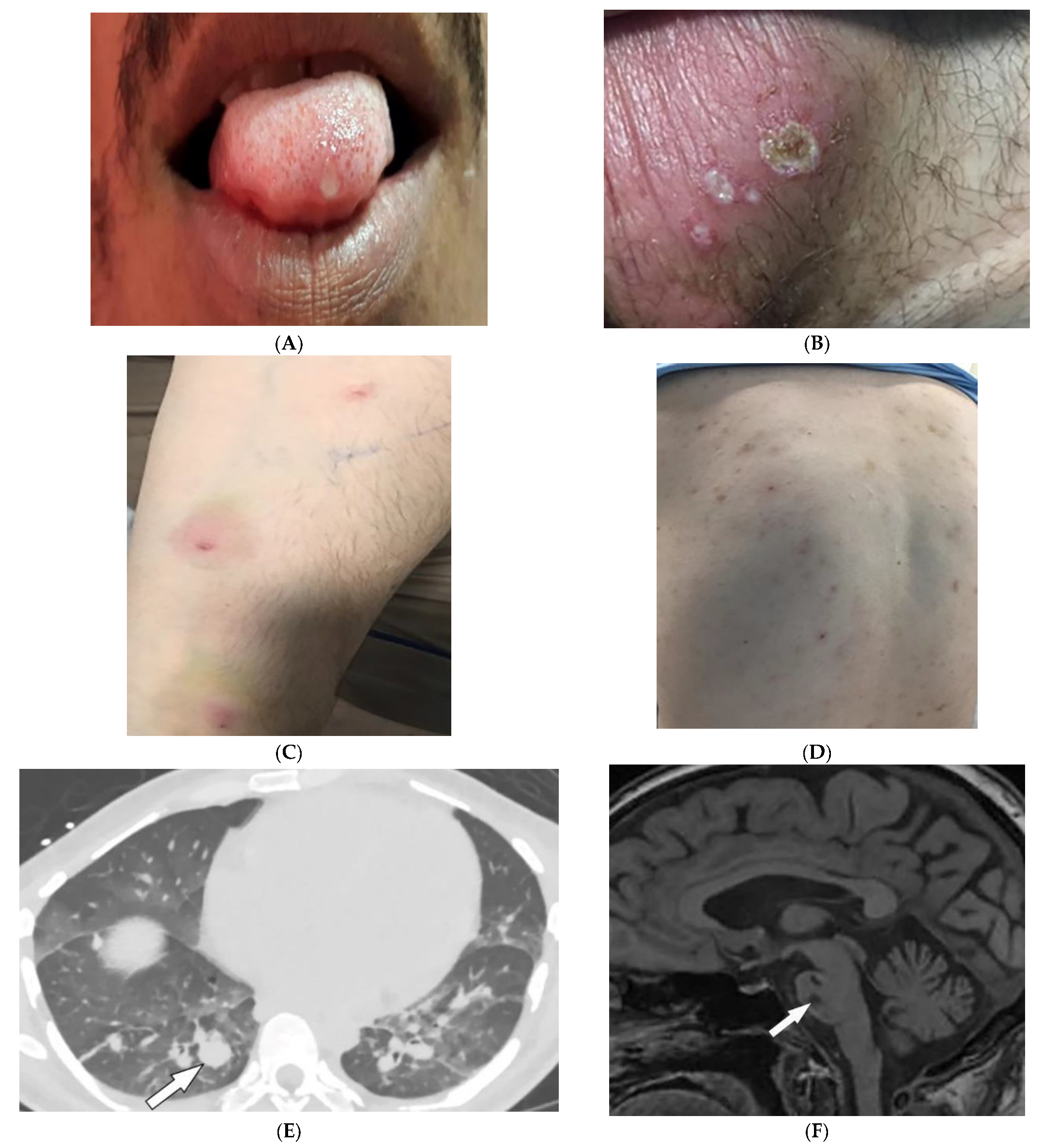
Figure 7. Dermatologic and radiologic images illustrative of Behcet’s disease. (A) Ulceration on the tongue of a 52-year-old male. (B) Scrotal ulcerations and erosions (C) Image of the forearm of a 35-year-old male showing a positive pathergy test. (D) Acneiform eruption with residual post-inflammatory hyperpigmentation. (E) Axial HRCT of the chest with contrast in the lung window shows multiple bilateral pulmonary artery aneurysms (arrow). (F) Sagittal FLAIR MRI of the brain displays pontine involvement in neuro-Behçet’s disease (arrow).
Vascular involvement in BD includes venous and arterial occlusion and aneurysmal dilation involving the abdominal aorta and pulmonary arteries. Early-stage findings include irregular wall thickening, perivascular fat stranding, and delayed mural enhancement, whereas late-stage features of arterial vasculitis include stenosis and aneurysmal formation on CT and MRI. Multiple, bilateral pulmonary artery aneurysms are a rare but characteristic feature of BD (Figure 7E), which may present as parahilar nodular opacities on chest radiograph. CTA/MRA delineate vessels and collaterals, the presence of thrombus, and evidence of mediastinal involvement [37][40]. CNS disease manifests in 10–50% of patients. Acute attacks initially involve the basal ganglia or brainstem, with extension to the diencephalic structures, and show contrast enhancement and scattered areas of T2 hyperintensity (Figure 7F). Months later, small scattered hyperintense lesions present in the periventricular white matter [37][40].
References
- Kolasinski, S.L.; Chi, A.S.; Lopez-Garib, A.J. Current Perspectives on Imaging for Systemic Lupus Erythematosus, Systemic Sclerosis, and Dermatomyositis/Polymyositis. Rheum. Dis. Clin. N. Am. 2016, 42, 711–732.
- Mainetti, C.; Terziroli Beretta-Piccoli, B.; Selmi, C. Cutaneous Manifestations of Dermatomyositis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2017, 53, 337–356.
- Ahuja, J.; Arora, D.; Kanne, J.P.; Henry, T.S.; Godwin, J.D. Imaging of Pulmonary Manifestations of Connective Tissue Diseases. Radiol. Clin. N. Am. 2016, 54, 1015–1031.
- Kamperman, R.G.; van der Kooi, A.J.; de Visser, M.; Aronica, E.; Raaphorst, J. Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review. Int. J. Mol. Sci. 2022, 23, 4301.
- Ran, J.; Dai, B.; Liu, C.; Zhang, H.; Li, Y.; Hou, B.; Li, X. The diagnostic value of T2 map, diffusion tensor imaging, and diffusion kurtosis imaging in differentiating dermatomyositis from muscular dystrophy. Acta Radiol. 2022, 63, 467–473.
- Kubinova, K.; Dejthevaporn, R.; Mann, H.; Machado, P.M.; Vencovsky, J. The role of imaging in evaluating patients with idiopathic inflammatory myopathies. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S114), 74–81.
- Schulze, M.; Kötter, I.; Ernemann, U.; Fenchel, M.; Tzaribatchev, N.; Claussen, C.D.; Horger, M. MRI Findings in Inflammatory Muscle Diseases and Their Noninflammatory Mimics. Am. J. Roentgenol. 2009, 192, 1708–1716.
- Smitaman, E.; Flores, D.V.; Gómez, C.M.; Pathria, M.N. MR Imaging of Atraumatic Muscle Disorders. Radiogr. A Rev. Publ. Radiol. Soc. N. Am. Inc. 2018, 38, 500–522.
- Belperio, J.A.; Shaikh, F.; Abtin, F.G.; Fishbein, M.C.; Weigt, S.S.; Saggar, R.; Lynch, J.P. Diagnosis and treatment of pulmonary sarcoidosis: A review. JAMA 2022, 327, 856–867.
- Guidry, C.; Fricke, R.G.; Ram, R.; Pandey, T.; Jambhekar, K. Imaging of Sarcoidosis: A Contemporary Review. Radiol. Clin. N. Am. 2016, 54, 519–534.
- Ganeshan, D.; Menias, C.O.; Lubner, M.G.; Pickhardt, P.J.; Sandrasegaran, K.; Bhalla, S. Sarcoidosis from Head to Toe: What the Radiologist Needs to Know. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2018, 38, 1180–1200.
- Lew, P.P.; Ngai, S.S.; Hamidi, R.; Cho, J.K.; Birnbaum, R.A.; Peng, D.H.; Varma, R.K. Imaging of Disorders Affecting the Bone and Skin. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2014, 34, 197–216.
- Redissi, A.; Penmetsa, G.K.; Litaiem, N. Lupus pernio. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Noe, M.H.; Rosenbach, M. Cutaneous sarcoidosis. Curr. Opin. Pulm. Med. 2017, 23, 482–486.
- Koyama, T.; Ueda, H.; Togashi, K.; Umeoka, S.; Kataoka, M.; Nagai, S. Radiologic Manifestations of Sarcoidosis in Various Organs. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2004, 24, 87–104.
- Sève, P.; Pacheco, Y.; Durupt, F.; Jamilloux, Y.; Gerfaud-Valentin, M.; Isaac, S.; Boussel, L.; Calender, A.; Androdias, G.; Valeyre, D.; et al. Sarcoidosis: A clinical overview from symptoms to diagnosis. Cells 2021, 10, 766.
- Ruaro, B.; Confalonieri, P.; Santagiuliana, M.; Wade, B.; Baratella, E.; Kodric, M.; Berria, M.; Jaber, M.; Torregiani, C.; Bruni, C.; et al. Correlation between Potential Risk Factors and Pulmonary Embolism in Sarcoidosis Patients Timely Treated. J. Clin. Med. 2021, 10, 2462.
- Barreras, P.; Stern, B.J. Clinical features and diagnosis of neurosarcoidosis–review article. J. Neuroimmunol. 2022, 368, 577871.
- Chapin, R.; Hant, F.N. Imaging of scleroderma. Rheum. Dis. Clin. N. Am. 2013, 39, 515–546.
- Kowalska-Kępczyńska, A. Systemic Scleroderma—Definition, Clinical Picture and Laboratory Diagnostics. J. Clin. Med. 2022, 11, 2299.
- Abbas, L.F.; O’Brien, J.C.; Goldman, S.; Pezeshk, P.; Chalian, M.; Chhabra, A.; Jacobe, H.T. A Cross-sectional Comparison of Magnetic Resonance Imaging Findings and Clinical Assessment in Patients With Morphea. JAMA Dermatol. 2020, 156, 590–592.
- Ruaro, B.; Sulli, A.; Pizzorni, C.; Paolino, S.; Smith, V.; Alessandri, E.; Trombetta, A.C.; Alsheyyab, J.; Cutolo, M. Correlations between blood perfusion and dermal thickness in different skin areas of systemic sclerosis patients. Microvasc. Res. 2018, 115, 28–33.
- Sulli, A.; Ruaro, B.; Cutolo, M. Evaluation of blood perfusion by laser speckle contrast analysis in different areas of hands and face in patients with systemic sclerosis. Ann. Rheum. Dis. 2014, 73, 2059–2061.
- Dzwigala, M.; Sobolewski, P.; Maslinska, M.; Yurtsever, I.; Szymanska, E.; Walecka, I. High-resolution ultrasound imaging of skin involvement in systemic sclerosis: A systematic review. Rheumatol. Int. 2021, 41, 285–295.
- Hughes, M.; Bruni, C.; Cuomo, G.; Delle Sedie, A.; Gargani, L.; Gutierrez, M.; Lepri, G.; Ruaro, B.; Santiago, T.; Suliman, Y.; et al. The role of ultrasound in systemic sclerosis: On the cutting edge to foster clinical and research advancement. J. Scleroderma Relat. Disord. 2021, 6, 123–132.
- Chatzinikolaou, S.L.; Quirk, B.; Murray, C.; Planche, K. Radiological findings in gastrointestinal scleroderma. J. Scleroderma Relat. Disord. 2020, 5, 21–32.
- Abenavoli, L.; Dastoli, S.; Bennardo, L.; Boccuto, L.; Passante, M.; Silvestri, M.; Proietti, I.; Potenza, C.; Luzza, F.; Nisticò, S.P. The Skin in Celiac Disease Patients: The Other Side of the Coin. Medicina 2019, 55, 578.
- Tennyson, C.A.; Semrad, C.E. Small bowel imaging in celiac disease. Gastrointest. Endosc. Clin. N. Am. 2012, 22, 735–746.
- Sheedy, S.P.; Barlow, J.M.; Fletcher, J.G.; Smyrk, T.C.; Scholz, F.J.; Codipilly, D.C.; Al Bawardy, B.F.; Fidler, J.L. Beyond moulage sign and TTG levels: The role of cross-sectional imaging in celiac sprue. Abdom. Radiol. 2017, 42, 361–388.
- Paolantonio, P.; Tomei, E.; Rengo, M.; Ferrari, R.; Lucchesi, P.; Laghi, A. Adult celiac disease: MRI findings. Abdom. Imaging 2007, 32, 433–440.
- Marzano, A.V.; Raimondo, M.G.; Berti, E.; Meroni, P.L.; Ingegnoli, F. Cutaneous Manifestations of ANCA-Associated Small Vessels Vasculitis. Clin. Rev. Allergy Immunol. 2017, 53, 428–438.
- Singhal, M.; Gupta, P.; Sharma, A. Imaging in small and medium vessel vasculitis. Int. J. Rheum. Dis. 2019, 22 (Suppl. S1), 78–85.
- Schmidt, W.A. Imaging in vasculitis. Best Pract. Res. Clin. Rheumatol. 2013, 27, 107–118.
- Howard, T.; Ahmad, K.; Swanson, J.A.; Misra, S. Polyarteritis nodosa. Tech. Vasc. Interv. Radiol. 2014, 17, 247–251.
- Chasset, F.; Frances, C. Cutaneous Manifestations of Medium- and Large-Vessel Vasculitis. Clin. Rev. Allergy Immunol. 2017, 53, 452–468.
- Diaz-Perez, J.L.; De Lagran, Z.M.; Diaz-Ramon, J.L.; Winkelmann, R.K. Cutaneous polyarteritis nodosa. Semin. Cutan. Med. Surg. 2007, 26, 77–86.
- Mehdipoor, G.; Davatchi, F.; Ghoreishian, H.; Arjmand Shabestari, A. Imaging manifestations of Behcet’s disease: Key considerations and major features. Eur. J. Radiol. 2018, 98, 214–225.
- Elbendary, A.; Abdel-Halim, M.R.; Ragab, G. Updates in cutaneous manifestations of systemic vasculitis. Curr. Opin. Rheumatol. 2022, 34, 25–32.
- Chae, E.J.; Do, K.-H.; Seo, J.B.; Park, S.H.; Kang, J.-W.; Jang, Y.M.; Lee, J.S.; Song, J.-W.; Song, K.-S.; Lee, J.H.; et al. Radiologic and Clinical Findings of Behçet Disease: Comprehensive Review of Multisystemic Involvement. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2008, 28, e31.
- Vural, S.; Boyvat, A. The skin in Behçet’s disease: Mucocutaneous findings and differential diagnosis. JEADV Clin. Pract. 2022, 1, 11–20.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
30 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No