Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elsa Garza-Treviño | -- | 2577 | 2022-08-29 16:32:07 | | | |
| 2 | Amina Yu | + 3 word(s) | 2580 | 2022-08-30 03:13:44 | | | | |
| 3 | Amina Yu | -9 word(s) | 2571 | 2022-08-31 08:16:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Islas, J.F.; Quiroz-Reyes, A.G.; Delgado-Gonzalez, P.; Franco-Villarreal, H.; Delgado-Gallegos, J.L.; Garza-Treviño, E.N.; Gonzalez-Villarreal, C.A. Cancer Stem Cells in Tumor Microenvironment. Encyclopedia. Available online: https://encyclopedia.pub/entry/26629 (accessed on 27 July 2026).
Islas JF, Quiroz-Reyes AG, Delgado-Gonzalez P, Franco-Villarreal H, Delgado-Gallegos JL, Garza-Treviño EN, et al. Cancer Stem Cells in Tumor Microenvironment. Encyclopedia. Available at: https://encyclopedia.pub/entry/26629. Accessed July 27, 2026.
Islas, Jose Francisco, Adriana G. Quiroz-Reyes, Paulina Delgado-Gonzalez, Hector Franco-Villarreal, Juan Luis Delgado-Gallegos, Elsa N. Garza-Treviño, Carlos A. Gonzalez-Villarreal. "Cancer Stem Cells in Tumor Microenvironment" Encyclopedia, https://encyclopedia.pub/entry/26629 (accessed July 27, 2026).
Islas, J.F., Quiroz-Reyes, A.G., Delgado-Gonzalez, P., Franco-Villarreal, H., Delgado-Gallegos, J.L., Garza-Treviño, E.N., & Gonzalez-Villarreal, C.A. (2022, August 29). Cancer Stem Cells in Tumor Microenvironment. In Encyclopedia. https://encyclopedia.pub/entry/26629
Islas, Jose Francisco, et al. "Cancer Stem Cells in Tumor Microenvironment." Encyclopedia. Web. 29 August, 2022.
Copy Citation
Cancer is a multi-step process during which cells acquire an untamed ability to grow, proliferate, and (most times) differentiate, ultimately leading to either improper organ growth or the establishment of inadequate cells in locations where they contribute negatively to the body homeostasis, causing (amongst other things) high levels of inflammation. Colorectal and gastric cancers are the most prevalent cancer in the digestive track. They represent the third and fifth leading causes of cancer-related deaths. With over 500,000–990,000 new cases worldwide, they typically have a five-year survival rate (mainly due to late diagnosis).
cancer stem cells
gastrointestinal adenocarcinomas
colon cancer and gastric cancer
1. Generalities and Risk Factors by Gastrointestinal Cancers
Most colorectal tumors develop slowly through a series of morphological, histological, genetic, and epigenetic changes. They are generally asymptomatic until they reach a considerable size, thus increasing the risk for adenomas to develop into colorectal cancer (CRC) [1][2]. Several factors increase the risk of developing this disease. Among the most studied were a red and/or processed meat diet, high alcohol intake, tobacco use, and sedentarism [3]. Other risk factors associated with the development of CRC are non-modifiable, such as a family history of colorectal polyps, CRC, lynch syndrome, inflammatory bowel disease, type 2 diabetes, and racial and ethnic backgrounds [4]. CRC typically develops from focal changes within benign, precancerous polyps. These polyps are localized growths or aggregations of abnormal cells within the intestinal mucosa that protrude into the intestinal lumen [4].
Whether sporadic or hereditary, gene mutations also increase the risk for CRC. Some mutations in DNA mismatch repair (MMR) genes, such as MLH1, MSH2, PMS2, and adenomatous polyposis coli gene (APC), are uncommon and have a low prevalence in hereditary cancer [5]. The chromosomal instability pathway is observed in 65–70% of sporadic cancers. The first mutations develop within the APC gene cell division and the next mutations develop in the KRAS oncogene, which affects cell differentiation, growth, motility, and survival. This causes a loss of function of the p53 gene, which acts as a transcription and apoptosis regulator that results in carcinogenesis [6]. Another epigenetic alteration seen in SSP-based CRC is aberrant gene promoter region hypermethylation, which inhibits gene transcription, regulating growth-promoting genes, bone morphogenic protein 3, and n-Myc downstream-regulated gene 4 (NDRG4) [4][6][7].
Common risk factors for gastric cancer (GC) include older age, male sex, tobacco smoking, radiation, and family history. Other factors are obesity, helicobacter pylori infection, gastroesophageal reflux disease, and diet (such as a high intake of salty foods or low vitamin A and C intake), nitrosamines, chemicals, smoked food, and high alcohol consumption [8]. In particular, high salt intake and tobacco are associated with increased incidence and mortality of GC [9]. The carcinomas are a consequence of a multistep process starting from chronic inflammatory through a sequence of atrophic gastritis, metaplasia, low-/high-grade dysplasia, and finally cancer. Gastritis is caused by chronic inflammation and is commonly regarded as a point of no return for carcinogenesis [10]. This results from persistent infection, which may evolve chronic atrophy gastritis, and subsequent changes in the gastric mucosa. Helicobacter pylori is the most prevalent bacterial pathogen currently classified as a group one carcinogen by the World Health Organization (definite carcinogen) [11].
2. Cancer Stem Cells and Tumoral Microenvironment
cancer stem cell (CSC) exhibit several remarkable abilities mentioned elsewhere. However, since they can derive from normal stem cells, they might also arise through dedifferentiation of mature somatic cells to reacquire stem cell characteristics. Studies have shown that CSCs are also responsible for therapy resistance by developing an increased expression of drug transporters, maintaining a slow dividing state, and efficient DNA damage repair mechanisms [12]. The identification of CSCs is based on markers found in normal stem cells, and these vary according to the type of cancer. CD44, CD24, and CD133 are usually common in gastrointestinal cancers. NANOG, OCT4, SOX2, and KLF4 transcription factors are considered critical of stem self-renewal and pluripotency regulators, mediating tumor proliferation and differentiation [10]. Also, overexpression of components from signaling pathways such as JAK/STAT, WNT, NOTCH, SHH, PI3K/phosphatase and Tensin homolog, and NF-κB signaling pathways are involved in renewal, differentiation, and uncontrolled proliferation of CSC [13].
Inside the tumor, the CSC niche can be a product of numerous factors specific to the TME and considered important factors extrinsically influencing tumor heterogeneity. These CSC can affect the tumor’s aggressiveness or invasion, modulating the normal growth and development of resident stem cells [14][15]. In turn, the accumulation of key mutations appears to begin specifically within the CSC compartment. Related EMT markers TGF-β1, TWIST, SNAIL, SLUG, vimentin, and CD44 were upregulated, and CDH1 mRNA levels were decreased in the gastric mucosa of patients with dysplasia or early GC in comparison with controls. A variant of CD44, a hyaluronic acid receptor (CD44v8-10), was identified as the predominant CD44 variant expressed in GC cells and contributes to tumor progression, possibly by enhancing oxidative stress defense [14].
Another proposed origin of CSCs is derived from the transformation of bone marrow-derived mesenchymal stem cells (BM-MSCs) in the GC, which served as primary cell components contributing to tumor progress, migration, and angiogenesis [16]. It has been observed that after infection and inflammatory stimulation of BM-MSCs, they migrate to the gastric epithelium and participate in tissue repair of the gastric mucosa [17].
A tumor’s diverse cell population tends to be erratic rather than regulated. However, CSC within the tumor is exposed to several biomolecules lead to an imbalance between CSC self-renewal and differentiation [17]. In CRC, MSC-derived exosomes are a double-edged sword in cancer development, metastasis, and invasion [18][19]. Tumor-associated stromal cells are key contributors to the tumor microenvironment, these arise from distinct sources, consisting of the basement membrane, non-malignant cells (fibroblasts, BM-MSC, adipocytes), immune cells (macrophages, lymphocytes), extracellular matrix, and vasculature (endothelial cells, pericytes) [20].
3. Crosstalk between Oncogenic Signaling and Metabolic Pathways of CSC: Role of Stroma-Derived Chemokines in the Local Invasion of Primary Tumor
Inflammation and tissue damage attract cells that cooperate with tumor TME as immune cells, stromal cells, adipocytes, and ECM components, whose main role is inhibiting apoptosis, protection of tumoral cells, promotion of proliferation, immune evasion, and invasion [21][22]. ECM is formed by a variety of substances, including collagen, elastin, laminin, fibronectin, and modulators such as metalloproteinase (MMP), which cleaves the ECM components and is crucial for tissue remodeling [23]. The persistent stimulus of inflammation in the tissue affects the homeostasis of cells, matrix, and cytokines. This finally leads to fibrosis and remodeling of ECM, which contributes to the establishment of tumors [24].
One example of this process is when the GC microenvironment becomes infiltrated by CAFs and TAMs, leading to excessive fibrosis [25]. The stimulus broadens the range of proinflammatory cytokines such as IL-6, TGF-β, FGF (fibroblast growth factor), TNF-α, and IL-1β that promote EMT [26]. Also, these cells induce fibrosis in ECM, which is associated with a worse prognosis in GC. In colitis-associated cancer, Liang et al. demonstrated that sphin-gosine-1-phosphate (S1P) induces an amplifying loop of SIPR1 and NF-kβ/IL-6/STAT3 [27]. Interleukins such as IL-6 and gp130-related are the main activators of the JAK2/STAT3 pathway in CSCs. In addition, IL-6 promotes the survival of pre-malignant intestinal epithelial cells, which then transforms into tumoral cells [28][29]. CAFs enhance GC cell migration and EMT through the secretion of IL-6 [25]. Once activated, the STAT-3 signaling pathway induces Tlr2 gene transcription in the gastric epithelium, which after overexpression, promotes proliferation and inhibits gastric epithelial cell apoptosis [30]. Tumor cells take advantage of STAT-3 by increasing immune evasion by inhibiting the maturation of dendritic cells (DC), which activate the adaptive immune response [31]. In recent years, WNT5a signaling in CAF has been implicated in tumor progression. In turn, overexpression of β-catenin and WNT5a indicates a poor prognosis since they promote cell growth, migration, invasion, and EMT of digestive tract tumors [32]. In addition, CSCs promote tumor infiltration of TAM through the CD44 receptor, which is upregulated by miR-328 suppression. Moreover, MSCs present an immunomodulatory role on lymphocytes B, T, dendritic cells, macrophages, and MDSCs. Therefore, MSC could affect CD4+ T cell migration and differentiation, T helper 17 homeostasis modulation, and response [26]. In addition, STAT3 mediated TWIST expression and EMT can be activated by EGF/EGFR [27].
Chemokines act by interacting with specific G protein-coupled receptors, and chemokines from TME can facilitate tumor progression or remodeling of the tumor niche by signal transduction [33]. Thus, these proteins may play a crucial role in the pathogenesis of CRC, and GC. However, the high heterogeneity of the cell context limits transcription factors potential to induce gene expression on CSCs and their changes to improve tumor progression [34]. It is being proposed that transcription factors such as the Yes-Associated Protein (YAP)/Transcriptional Co-activator with PDZ-binding Motif (TAZ) pathway lead to epithelial phenotype repression and participate together with factors of the SNAIL family, such as TWIST and Zeb, which are the main regulators of EMT [35]. YAP overexpression has been reported in GC, and CRC; protein levels are associated with poor prognosis, tumor stage, and metastasis [36][37][38].
Inflammation and tissue damage are a potent chemokine source; both processes recruit cells that cooperate with tumor TME as immune cells, stromal cells, adipocytes, and ECM components. The main role of these cells is to inhibit apoptosis, protect tumoral cells, promote proliferation, and immune evasion and invasion [21][22].
Gastrointestinal epithelial cell infection by H. pylori and the expression of CXCL1 gastric cancer cells is necessary to stimulate the migration of bone marrow-derived stromal cells (BMDC) by CXCR2 signaling; moreover, the expression of CD271 by BMDC is related with invasion and worse prognosis [39]. Tumor-associated macrophages (TAM) are associated with tumor stage and metastasis. There are two types of macrophage differentiation: M1 macrophages that produce type I proinflammatory cytokines such as IL-1β, IL-1α, IL-12, TNF-α, and GFAP, and M2 macrophages that produce type II anti-inflammatory cytokines such as IL-4, IL-6, and IL-10, related to the pro-tumorigenic activity. The change from one phenotype to another depends on the TME, and the high infiltration of M2 is related to a worse prognosis [40]. Chronic gastritis induced by H. pylori infection is associated with 90% of gastric cancer cases. It has been reported that this infection causes atrophy of acid-secreting parietal cells (PC), which increases CD44+ stem cell proliferation in the gastric isthmus by pERK [9][41]. Some gastrointestinal hormones, such as gastrin or neurotransmitters and acetylcholine (Ach), may also play unique roles in the antral stem cell niche. Gastrin is secreted from G cells and is responsible for HCl secretion in parietal cells (PCs). These cells reside near the antral isthmus region [42]. Gastrin also induces the expression of EGFR ligands as heparin-binding EGF and trefoil family factor 2, which activate the cholecystokinin receptor (CCK-BR), and thus, motility, secretion, migration, and proliferation of gastric cells [43]. Moreover, MMPs and tissue inhibitors of metalloproteinases (TIMPs) decrease E-Cadherin and ECM degradation cell change interactions and paracrine signals, helping malignant transformation [44]. For example, H. pylori increase IL-21 expression in infected gastric mucosa and promote gelatinases, MMP-2, MMP-7, and MMP-9 synthesis through NF-kβ in activated B cells [44]. Some other proteins increase their expression during GC, such as phosphoglycerate kinase 1 (PGK1), CXCR4, CXCL12, and β-catenin [45], which promote EMT. In addition, CAFs secrete multiple proinflammatory cytokines such as TGF-β1, IL-β1, IL-6, IL-33, ROS, C-X-C chemokine receptor (CXC), MMPs, lysyl oxidase, miR-21, TNF-α, and αSMA [45]. All of these factors contribute to tissue fibrosis, and later fibrogenesis activates ECM remodeling. In addition, fibrogenesis activates the chemoresistance-inducing factor SNAIL in epithelial cells, promoting proliferation and inducing drug resistance [46].
4. Participation of Extracellular Matrix Components in Cancer Progression
The role of the ECM has been demonstrated in all stages of GC and CRC, from onset to metastasis [47][48]. However, it has been shown that some components of the protein family are shared by gastrointestinal tumors and regulate a key aspect in the early stages of tumor biology, participating in the regulation of CSC (which can be favored under the stimulation of some signs or changes in the microenvironment by ECM proteins) [49]. Intestinal epithelial cells express integrins in healthy and pathological circumstances. Integrins are categorized based on the preferred ligands; they bind to collagens for α1/α2 coupled to β1, laminins for α3/α6/α7 coupled to β1 or β4, tenascin for α9β1, and RGD-containing ligands (fibronectin, osteopontin, and vitronectin) for α5/α8 coupled to β1 and αV coupled to β1/β3/β5/β6/β8. Recent works address the altered expression of certain integrins and their involvement in GC and CRC progression [50]. For example, integrin V6 can be expressed by CRC cells, which can then inactivate TGFβ by integrin αvβ6 subsequently activating fibroblasts that promote tumor invasion [51]. CRC integrin αvβ8 expressed on tumor cells is reported as a crucial regulatory function during cell adhesion in the tumor microenvironment and supports the activation of TGF-β1 [52]. Several of these integrins, including integrin subunits β1, α6, β3, and β4, concentrated in healthy adult stem and progenitor cells, are also signs of CSCs. Another characteristic of CRC is mainly overexpression of collagen types I, VI, VII, VIII, X, XI, and XVIII. For example, increased expression of type 1 collagen promotes EMT and stem cell marker expression by activation of PI3K/AKT/Snail signaling pathway conducted by integrin α2β1 [53]. In addition, recent studies reported a higher expression of collagen XVII, which was significantly associated with the progression of cancer and by interaction with laminin-5 (Laminin-332), which is essential for epithelial cell migration and basement membrane attachment and, according to some studies, is a determinant for CCR initiation [54]. Figueiredo et al. reported that mutant E-cadherin conduces to an increase in β1 integrin expression associated with higher grade tumors and reduced overall survival of the GC patient [55]. Some of these proteins promote key CSC functions such as: (1) EMT; (2) immune surveillance modulation; (3) self-renewal/maintenance; and (4) metabolic reprogramming and matricellular proteins. These functions are also involved in the cellular mechanical response, such as mechano-sensor integrins, which are receptors for many proteins [56].
Another important component in ECM are miRNAs. miR-206 downregulation in GC enhances GC stem cells (GCSCs) and carcinogenesis. At the same time, its overexpression suppresses GCSCs formation and associated tumorigenesis through ETS homologous factor (EHF) downregulation [57]. In GC, miR-17-92 members can help show metaplasia, but more importantly, they can be found at the early pre-niche stage serving a significant role as biomarkers [58]. In CRC, one of the most studied pathologies, circulating levels of miR-17 and miR-92a have been associated with early stages (primary tumor establishment) formation [59][60]. Having members of this cluster circulating (if further analyzed and quantified) can give us an idea of cancer staging in the gastrointestinal tract. Moreover, miR-340 is a well-studied miRNA known for its role in the early establishment of CRC [61][62]. The primary miRNAs that participate in several pathways and lead to or inhibit gastric and colon cancers are shown in Figure 1.
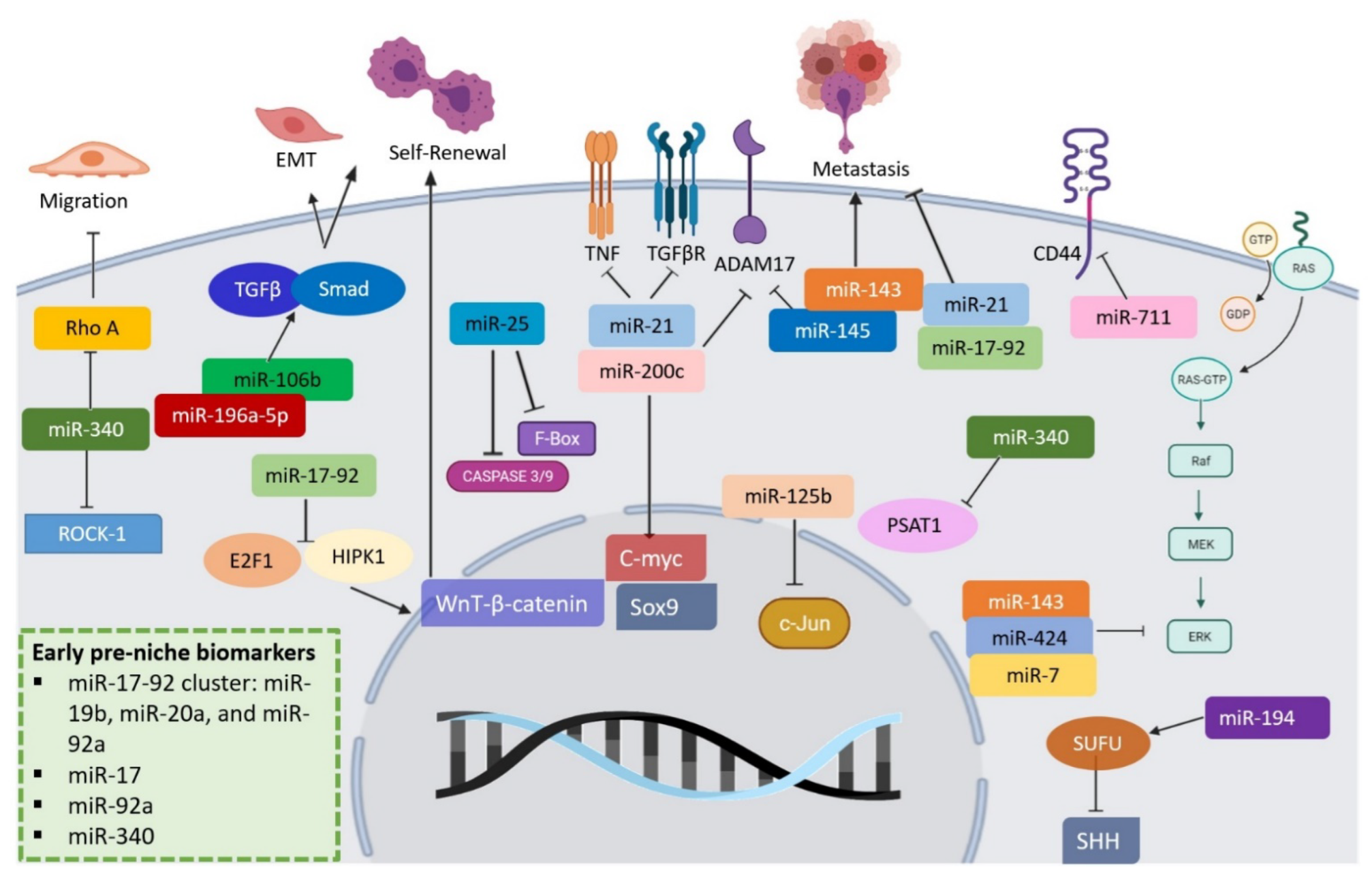
Figure 1. Regulatory roles of miRNAs in gastrointestinal cancers. miRNAs participate in the modification of several pathways that lead to or inhibit gastric and colon cancers. Some are present since cancer pre-niche and are related to EMT, migration, proliferation, invasion, and metastasis development.
In a bit more detail, in CD44 (+) cells, miR-106b enhances gastric stem cell (GCSC) traits such as EMT, self-renewal, and invasion through the modulation of the TGF-β/Smad signaling pathway [57]. miR-145 can further affect the stemness property of CD44 (+) cells, reducing levels of ADAM17 and SOX9, which regulate invasiveness. Loss of this regulation leads to tumor initiation via IL-6 inflammatory processes [63]. In addition, miR-196a-5p has been upregulated in CD44 + cells, and suppression led to less colony formation and invasion of GC stem cells, suggesting a significant role in EMT and invasion of this cell population [48]. miR-143, miR-145, miR-21, and miR-194 significantly distorted the epithelium of several cancers; these miRNAs relate to the target cell cycle (cdc25), PTEN signaling, FSCN1, ZEB2, and MDM2 [64][65][66][67][68].
References
- Lochhead, P.; Chan, A.T.; Giovannucci, E.; Fuchs, C.S.; Wu, K.; Nishihara, R.; O’brien, M.; Ogino, S. Progress and Opportunities in Molecular Pathological Epidemiology of Colorectal Premalignant Lesions. Am. J. Gastroenterol. 2014, 109, 1205.
- Conteduca, V.; Sansonno, D.; Russi, S.; Dammacco, F. Precancerous colorectal lesions (Review). Int. J. Oncol. 2013, 43, 973–984.
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103.
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976.
- De’angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072.
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am. J. Surg. Pathol. 2006, 30, 1491–1501.
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric Cancer: Descriptive Epidemiology, Risk Factors, Screening, and Prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713.
- Guggenheim, D.E.; Shah, M.A. Gastric cancer epidemiology and risk factors. J. Surg. Oncol. 2013, 107, 230–236.
- Xiao, S.; Zhou, L. Gastric Stem Cells: Physiological and Pathological Perspectives. Front. Cell Dev. Biol. 2020, 8, 571536.
- Ahn, H.J.; Lee, D.S. Helicobacter pylori in gastric carcinogenesis. World J. Gastrointest. Oncol. 2015, 7, 455–465.
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896.
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19.
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53.
- Zhao, Y.; Dong, Q.; Li, J.; Zhang, K.; Qin, J.; Zhao, J.; Sun, Q.; Wang, Z.; Wartmann, T.; Jauch, K.W.; et al. Targeting cancer stem cells and their niche: Perspectives for future therapeutic targets and strategies. Semin. Cancer Biol. 2018, 53, 139–155.
- Becerril-Rico, J.; Alvarado-Ortiz, E.; Toledo-Guzmán, M.E.; Pelayo, R.; Ortiz-Sánchez, E. The cross talk between gastric cancer stem cells and the immune microenvironment: A tumor-promoting factor. Stem Cell Res. Ther. 2021, 12, 498.
- Fu, Y.; Li, H.; Hao, X. The self-renewal signaling pathways utilized by gastric cancer stem cells. Tumor Biol. 2017, 39, 1010428317697577.
- Zhou, J.; Tan, X.; Tan, Y.; Li, Q.; Ma, J.; Wang, G. Mesenchymal stem cell derived exosomes in cancer progression, metastasis and drug delivery: A comprehensive review. J. Cancer 2018, 9, 3129–3137.
- Muralikumar, M. Current understanding of the mesenchymal stem cell-derived exosomes in cancer and aging. Biotechnol. Rep. 2021, 31, e00658.
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84.
- Kwilasz, A.J. The Tumor Microenvironment in Esophageal Cancer. Oncogene 2016, 35, 5337–5349.
- Han, P.; Cao, P.; Hu, S.; Kong, K.; Deng, Y.; Zhao, B.; Li, F. Esophageal microenvironment: From precursor microenvironment to premetastatic niche. Cancer Manag. Res. 2020, 12, 5857–5879.
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464.
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. DMM Dis. Model. Mech. 2011, 4, 165–178.
- Ham, I.H.; Lee, D.; Hur, H. Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J. Oncol. 2019, 2019.
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin. Cancer Biol. 2017, 47, 177–184.
- Li, B.; Huang, C. Regulation of EMT by STAT3 in gastrointestinal cancer (Review). Int. J. Oncol. 2017, 50, 753–767.
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and STAT3 are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113.
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217.
- Tye, H.; Kennedy, C.L.; Najdovska, M.; McLeod, L.; McCormack, W.; Hughes, N.; Dev, A.; Sievert, W.; Ooi, C.H.; Ishikawa, T.O.; et al. STAT3-Driven Upregulation of TLR2 Promotes Gastric Tumorigenesis Independent of Tumor Inflammation. Cancer Cell 2012, 22, 466–478.
- Su, Y.L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in tumor-associated myeloid cells: Multitasking to disrupt immunity. Int. J. Mol. Sci. 2018, 19, 1803.
- Qin, Y.; Wang, F.; Ni, H.; Liu, Y.; Yin, Y.; Zhou, X.; Gao, G.; Li, Q.; Qi, X.; Li, J. Cancer-associated fibroblasts in gastric cancer affect malignant progression via the CXCL12-CXCR4 axis. J. Cancer 2021, 12, 3011–3023.
- Pawluczuk, E.; Łukaszewicz-Zając, M.; Mroczko, B. The role of chemokines in the development of gastric cancer-diagnostic and therapeutic implications. Int. J. Mol. Sci. 2020, 21, 8456.
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157.
- Kim, H.; Lee, S.; Shin, E.; Seong, K.M.; Jin, Y.W.; Youn, H.S.; Youn, B.H. The Emerging Roles of Exosomes as EMT Regulators in Cancer. Cells 2020, 9, 861.
- Yin, F.; Dong, J.; Kang, L.; Liu, X. Hippo-YAP signaling in digestive system tumors. Am. J. Cancer Res. 2021, 11, 2495–2507.
- Qu, Y.; Zhang, L.; Wang, J.; Chen, P.; Jia, Y.; Wang, C.; Yang, W.; Wen, Z.; Song, Q.; Tan, B.; et al. Yes—Associated protein (YAP) predicts poor prognosis and regulates progression of esophageal squamous cell cancer through epithelial—Mesenchymal transition. Exp. Ther. Med. 2019, 18, 2993–3001.
- Jiang, L.; Zhang, J.; Xu, Q.; Wang, B.; Yao, Y.; Sun, L.; Wang, X.; Zhou, D.; Gao, L.; Song, S.; et al. YAP promotes the proliferation and migration of colorectal cancer cells through the Glut3/AMPK signaling pathway. Oncol. Lett. 2021, 21, 312.
- Kasashima, H.; Yashiro, M.; Nakamae, H.; Masuda, G.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; Sakurai, K.; Toyokawa, T.; et al. Bone marrow-derived stromal cells are associated with gastric cancer progression. Br. J. Cancer 2015, 113, 443–452.
- Wang, X.L.; Jiang, J.T.; Wu, C.P. Prognostic significance of tumor-associated macrophage infiltration in gastric cancer: A meta-analysis. Genet. Mol. Res. 2016, 15, gmr15049040.
- Khurana, S.S.; Riehl, T.E.; Moore, B.D.; Fassan, M.; Rugge, M.; Romero-Gallo, J.; Noto, J.; Peek, R.M.; Stenson, W.F.; Mills, J.C. The hyaluronic acid receptor CD44 coordinates normal and metaplastic gastric epithelial progenitor cell proliferation. J. Biol. Chem. 2013, 288, 16085–16097.
- Craven, C.J. A hypothesis of couplet molecules and couplet cells in gastric function and an association with Helicobacter pylori. BMC Gastroenterol. 2016, 16, 16.
- Smith, J.P.; Nadella, S.; Osborne, N. Gastrin and Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 75–83.
- Brücher, B.L.D.M.; Jamall, I.S. Transition from normal to cancerous cell by precancerous niche (PCN) induced chronic cell-matrix stress. 4Open 2019, 2, 14.
- Brücher, B.L.D.M.; Jamall, I.S. Precancerous niche (PCN), a product of fibrosis with remodeling by incessant chronic in flammation. 4 Open 2019, 2, 11.
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972.
- Fernandes, E.; Sores, J.; Cotton, S.; Peixoto, A.; Ferreira, D.; Freitas, R.; Reis, C.A.; Santos, L.L.; Ferreira, J.A. Esophageal, gastric and colorectal cancers: Looking beyond classical serological biomarkers towards glycoproteomics-assisted precision oncology. Theranostics 2020, 10, 4903–4928.
- Andreuzzi, E.; Capuano, A.; Poletto, E.; Pivetta, E.; Fejza, A.; Favero, A.; Doliana, R.; Cannizzaro, R.; Spessotto, P.; Mongiat, M. Role of extracellular matrix in gastrointestinal cancer-associated angiogenesis. Int. J. Mol. Sci. 2020, 21, 3686.
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86.
- Hou, S.; Wang, J.; Li, W.; Hao, X.; Hang, Q. Roles of Integrins in Gastrointestinal Cancer Metastasis. Front. Mol. Biosci. 2021, 8, 708779.
- Zhang, Z.; Vuori, K.; Reed, J.C.; Ruoslahti, E. The α5β1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Acad. Sci. USA 1995, 92, 6161–6165.
- Zhou, M.; Niu, J.; Wang, J.; Gao, H.; Shahbaz, M.; Niu, Z.; Li, Z.; Zou, X.; Liang, B. Integrin αvβ8 serves as a Novel Marker of Poor Prognosis in Colon Carcinoma and Regulates Cell Invasiveness through the Activation of TGF-B1. J. Cancer 2020, 11, 3803–3814.
- Wu, X.; Cai, J.; Zuo, Z.; Li, J. Collagen facilitates the colorectal cancer stemness and metastasis through an integrin/PI3K/AKT/Snail signaling pathway. Biomed. Pharmacother. 2019, 114, 108708.
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437.
- Figueiredo, J.; Ferreira, R.M.; Xu, H.; Gonçalves, M.; Barros—Carvalho, A.; Cravo, J.; Maia, A.F.; Carneiro, P.; Figueiredo, C.; Smith, M.L.; et al. Integrin β1 orchestrates the abnormal cell—Matrix attachment and invasive behaviour of E-cadherin dysfunctional cells. Gastric Cancer 2022, 25, 124–137.
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135.
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840.
- Li, H.; Wu, Q.; Li, T.; Liu, C.; Xue, L.; Ding, J.; Shi, Y.; Fan, D. The miR-17-92 cluster as a potential biomarker for the early diagnosis of gastric cancer: Evidence and literature review. Oncotarget 2017, 8, 45060–45071.
- Gonzalez-Villarreal, C.A.; Quiroz-Reyes, A.G.; Islas, J.F.; Garza-Treviño, E.N. Colorectal Cancer Stem Cells in the Progression to Liver Metastasis. Front. Oncol. 2020, 10, 1511.
- Fu, F.; Jiang, W.; Zhou, L. Circulating Exosomal miR-17-5p and miR-92a-3p Predict Pathologic Stage and Grade of Colorectal Cancer. Transl. Oncol. 2018, 11, 221–232.
- Algaber, A.; Al-Haidari, A.; Madhi, R.; Rahman, M.; Syk, I.; Thorlacius, H. MicroRNA-340-5p inhibits colon cancer cell migration via targeting of RhoA. Sci. Rep. 2020, 10, 16934.
- Sun, Y.A.N.; Zhao, X.; Zhou, Y.; Hu, Y.U. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol. Rep. 2012, 28, 1346–1352.
- Yu, C.-C.; Tsai, L.-L.; Wang, M.-L.; Yu, C.-H.; Lo, W.-L.; Chang, Y.-C.; Chiou, G.-Y.; Chou, M.-Y.; Chiou, S.-H. miR145 Targets the SOX9/ADAM17 Axis to Inhibit Tumor-Initiating Cells and IL-6–Mediated Paracrine Effects in Head and Neck Cancer. Cancer Res. 2013, 73, 3425–3440.
- Wu, X.; Ajani, J.A.; Gu, J.; Chang, D.W.; Tan, W.; Hildebrandt, M.A.T.; Huang, M.; Wang, K.Y.; Hawk, E. MicroRNA expression signatures during malignant progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer Prev. Res. 2013, 6, 196–205.
- Pack, S.D.; Karkera, J.D.; Zhuang, Z.; Pak, E.D.; Balan, K.V.; Hwu, P.; Park, W.S.; Pham, T.; Ault, D.O.; Glaser, M.; et al. Molecular cytogenetic fingerprinting of esophageal squamous cell carcinoma by comparative genomic hybridization reveals a consistent pattern of chromosomal alterations. Genes Chromosom. Cancer 1999, 25, 160–168.
- Shibuya, H.; Iinuma, H.; Shimada, R.; Horiuchi, A.; Watanabe, T. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology 2011, 79, 313–320.
- Wijnhoven, B.P.L.; Hussey, D.J.; Watson, D.I.; Tsykin, A.; Smith, C.M.; Michael, M.Z. MicroRNA profiling of Barrett’s oesophagus and oesophageal adenocarcinoma. Br. J. Surg. 2010, 97, 853–861.
- He, B.; Yin, B.; Wang, B.; Xia, Z.; Chen, C.; Tang, J. microRNAs in esophageal cancer (Review). Mol. Med. Rep. 2012, 6, 459–465.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
846
Revisions:
3 times
(View History)
Update Date:
31 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No