 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luis R. Domingo | + 1529 word(s) | 1529 | 2020-10-19 12:16:04 | | | |
| 2 | Catherine Yang | -507 word(s) | 1022 | 2020-10-20 03:01:47 | | |
Video Upload Options
The Molecular Electron Density Theory (MEDT) was proposed in 2016 as a contemporary reactivity model in Organic Chemistry against the Frontier Molecular Orbital (FMO) theory.
1. The Molecular Electron Density Theory
A Contemporary Reactivity Model in Organic Chemistry. The Molecular Electron Density Theory [1] (MEDT), which proposes that the changes in the electron density along the reaction path are responsible for the feasibility of an organic reaction, was proposed in 2016 by Luis R. Domingo after more than 300 published theoretical studies in the field of the Organic Chemistry. MEDT goes against any analysis based on molecular orbitals (MOs), such as the Fukui’s Frontier Molecular Orbital [2] (FMO) theory and those based in the Morokuma’s energy decomposition analysis.[3][4] These MO analyses were developed in the past century before the development of the quantum chemical tools allowing the topological analysis of the electron density.
2. History
Studying the competitive Diels-Alder (DA) [5] reactions and Michael [6] additions of dimethyl acetylenedicarboxylate (DMAD) with a series of 2-vinil pyrroles by using a simple AM1 semiempirical computational model, in 1995, Domingo found that both competitive reactions had very similar transition estate geometries (TSs).[7] The TSs for the DA reaction were found completely different to that proposed for the pericyclic mechanism proposed in 1969 by Woodward and Hoffmann.[8] An ab initio and DFT study for the reaction of DMAD with N-methyl pyrrole suggested a stepwise mechanism, in which the first step was shared by both reactions (see Scheme 1). [9]
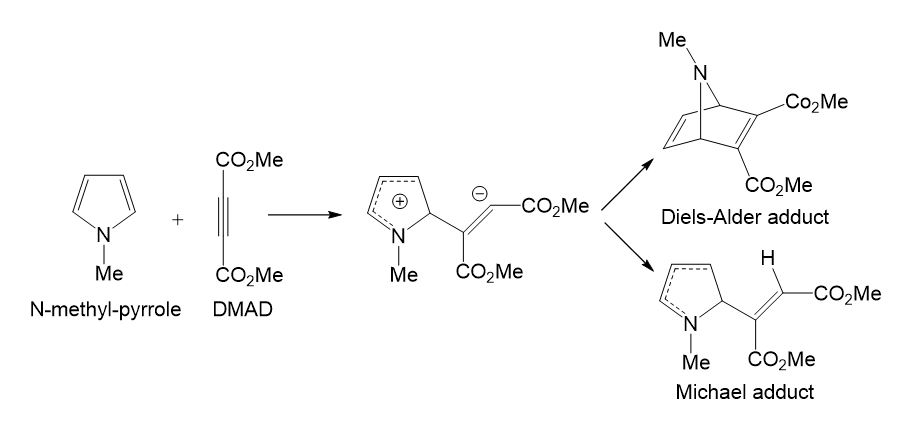
Scheme 1. Competitive DA and Michael reactions in the reaction of DMAD with N-methyl-pyrrole.
Further, in 1999, in a study of the DA reaction of nitroethene and a series of substituted ethylenes, Domingo established a correlation between the electrophilic and nucleophilic character of the reagents involved in these DA reactions and the polarity of the reaction measured by the global electron density transfer (GEDT) at the TSs.[10] Electrophilicity and nucleophilicity are concepts widely used in Organic Chemistry, developed in the first quarter of the 20th century. That study emphasized that the increase of the electrophilic and nucleophilic character of the reagents involved in these DA reactions decreased the reaction rates through more polar TSs.
The DA reactions between cyclopentadiene and the series of substituted cyano ethylenes, experimentally studied by Sauer in 1964 (see Scheme 2),[11] is a reference in many organic text books[12] showing how the electron-withdrawing substitution accelerates DA reactions. In 2003, Domingo studied the series of Sauer’s DA reactions,[13] finding two interesting conclusions: i) the reaction rate increases with the electrophilic character of the ethylene; and ii) the symmetric substitution in the ethylene decreases the effectiveness of the substitution. The geometries of the TSs, synchronous or highly asynchronous, were found completely dependent on the symmetry of the ethylenes. Non-symmetric substitution on the ethylene favours highly asynchronous TSs, which are associated to a two-center nucleophilic/electrophilic interaction.
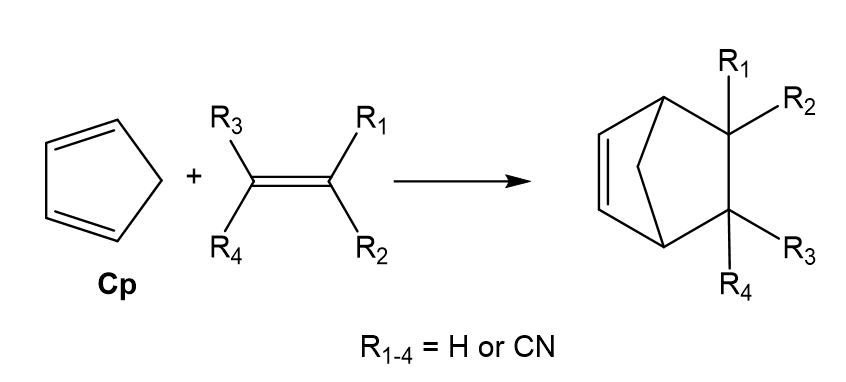
Scheme 2. Diels-Alder reactions between Cp and the cyanoethylene series.
After many studies of DA reactions, in 2009 Domingo proposed the polar Diels-Alder mechanism,[14] against the pericyclic one,[8] in which the favourable nucleophilic/electrophilic interactions taking place at the TSs control these relevant organic reactions. In that study, a complete linear correlation between the GEDT found at the TSs and the logarithm of the experimental reaction rate of Sauer’s series was established for the first time (see Figure 1).
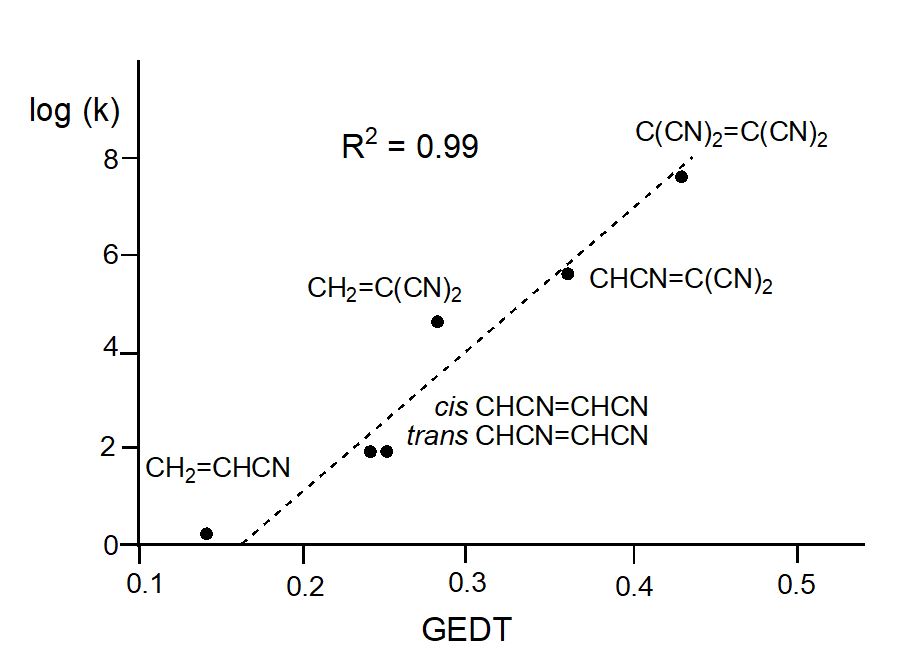
Figure 1. Plot of the logarithm of the experimental rate constant k vs GEDT, in average number of electrons, e, for the DA reactions of Cp with the cyanoethylene series; R2 = 0.99. Calculations were performed at the B3LYP/6-31G(d) level.
After many studies of reaction mechanisms based on the topological analysis of the Electron Localizarion Function (ELF), in 2014, Domingo published a review entitled “A new C–C bond formation model based on the quantum chemical topology of electron density”.[15] This review established that formation of the new C-C single bonds in cycloaddition reactions takes place by the C-to-C coupling of two pseudoradical centers[16] formed along the reaction paths, at a C-C distance of 2.0-1.90 Å. In the biography of this review it was wrote “while the distribution of the electron density is responsible for the molecular shape and physical properties, the capability for changes in electron density is responsible for the reactivity”, this being the foundation of the MEDT published later in 2016.[1]
[3+2] Cycloaddition (32CA) reactions are, together with DA reactions, one of the most important organic reactions. However, the three-atom-components (TACs) participating in 32CA reactions have different behaviours than those of dienes participation in DA reactions. After a huge work carried out within MEDT in the period 2015-2018, Domingo and Ríos-Gutiérrez recognised four different classes of TACs participating in 32CA reactions, i.e. pseudodiradical, pseudoradical, zwitterionic and carbenoid, proposing in 2019 the classification of 32CA reactions into four different types: pdr-type, pmr-type, zw-type and cb-type, respectively (see Scheme 3).[17] That MEDT studies allowed establishing a good correlation between the electronic structure of the simplest TACs and the reactivity type, allowing to explain the reactivity of the TACs and selectivity experimentally found in these important cycloaddition reactions.
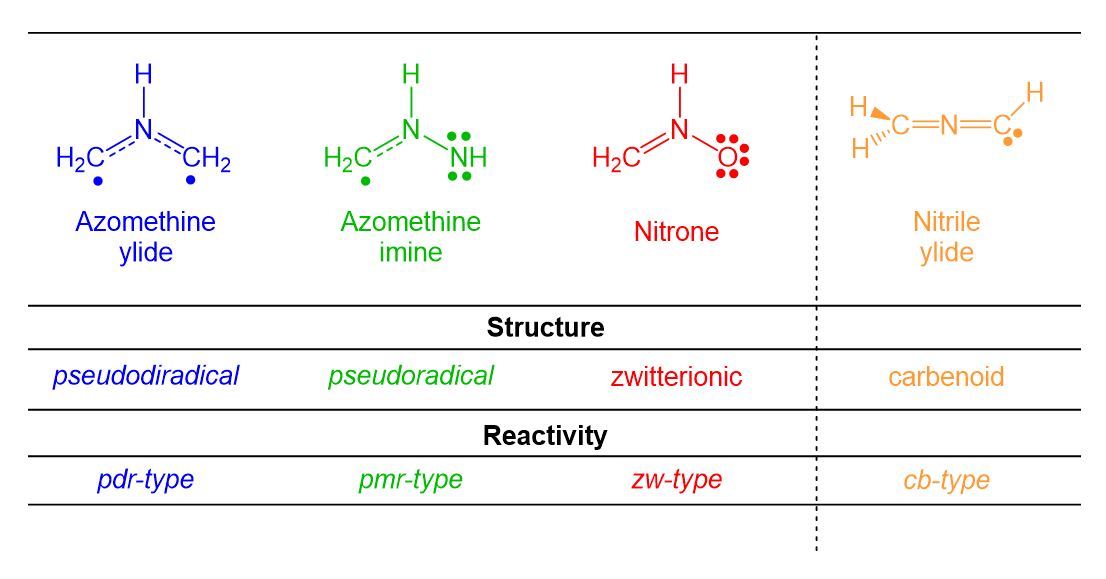
Scheme 3. Electronic structure of TACs and proposed reactivity types in 32CA reactions.
The intensive work carried out in the last 25 years in the field of the Organic Chemistry allowed Domingo establishing MEDT in 2016 as a modern theory for the study of organic reactivity against outdated theories and models based on the analysis of MOs established in the past century, and before the development of the quantum chemical tools able to characterize the electron density in organic molecules.
3. Perspective
Since the publication of MEDT in 2016, more than 150 theoretical studies have been developed within this theory, covering different organic reactions. This extensive work has proved that today the analysis of the electron density, which is a physical observable,[18][19] along a chemical reaction by using quantum chemical tools such as the analysis of the conceptual DFT (CDFT) indices,[20][21], the topological analysis of the electron density through Atoms in Molecules (AIM),[22], the topological analysis of the ELF,[23] and more recently the Non-Covalent Interactions (NCI),[24] permits the study of any aspect of the chemical organic reactivity, against the analyses based on MOs, which are not physical observables but only mathematical constructs that cannot be determined by experiment.[25]
References
- L. R. Domingo. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules, 2016, 21, 1319.
- K. Fukui. Molecular Orbitals in Chemistry, Physics, and Biology., New York, 1964.
- K. Morokuma, K. Kitaura. in Chemical Applications of Atomic and Molecular Electrostatic Potentials. Plenum: New York, 1981.
- F. M. Bickelhaupt. Understanding reactivity with Kohn–Sham molecular orbital theory: E2–SN2 mechanistic spectrum and other concepts. J. Comput. Chem., 1999, 20, 114–128.
- O. Diels, K. Alder. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98-122.
- A. Michael. Ueber die Addition von Natriumacetessig‐ und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren. J. Prakt. Chem., 1887, 35, 349-356.
- L. R. Domingo, R. A. Jones, M. T. Picher, J. Sepulveda-Arques. Theoretical Study of the Reaction of Dimethyl Acetylenedicarboxilate with 1-Methyl-2-(1-Substituted Vinyl) Pyrroles. Tetrahedron 1995, 51, 8739-8748.
- R. B. Woodward, R. Hoffmann. The Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781-853.
- L. R. Domingo, M. T. Picher, R. J. Zaragozá. Toward an Understanding of the Molecular Mechanism of the Reaction between 1-Methylpyrrole and Dimethyl Acetylenedicarboxylate. An ab Initio Study. J. Org. Chem. 1998, 63, 9183-9189.
- L. R. Domingo, M. Arnó, J. Andrés. Influence of Reactant Polarity on the Course of the Inverse-Electron-Demand Diels-Alder Reaction. A DFT Study of Regio- and Stereoselectivity, Presence of Lewis Acid Catalyst, and Inclusion of Solvent Effects in the Reaction between Nitroethene and Substituted Ethenes. J. Org. Chem. 1999, 64, 5867-5875.
- J. Sauer, H. Wiest, A. Mielert. Eine studie der Diels-Alder-reaktion, I. Die reaktivität von dienophilen gegenüber cyclopentadien und 9.10-dimethyl-anthracen. Chem. Ber. 1964, 97, 3183-3207.
- Carruthers, W. Some Modern Methods of Organic Synthesis. Cambridge University Press, Cambridge, 1978.
- L. R. Domingo, M. J. Aurell, P. Pérez, R .Contreras. Origin of the synchronicity on the transition structures of polar Diels-Alder reactions. Are these reactions [4 + 2] processes?. J. Org. Chem. 2003, 68, 3884-3890.
- L. R. Domingo, J. A. Sáez. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576-3583.
- L. R. Domingo. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415-32428.
- L. R. Domingo, J. A. Sáez. Understanding the Electronic Reorganization along the Nonpolar [3 + 2] Cycloaddition Reactions of Carbonyl Ylides. J. Org. Chem. 2011, 76, 373-379.
- M. Ríos-Gutiérrez, L. R. Domingo. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 267–282.
- P. Coppens, X-ray Charge Densities and Chemical Bonding. Oxford University Press: New York, 1997.
- T. S. Koritsanszky, P. Coppens. Chemical applications of X-ray charge-density analysis. Chem. Rev. 2001, 101, 1583-1628.
- R. G. Parr, W.Yang. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989.
- L. R. Domingo, M. Ríos-Gutiérrez, P. Pérez. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748.
- Bader, R. F. W. Atoms in Molecules. A Quantum Theory; Claredon Press, Oxford, U.K, 1990.
- A. D. Becke, K. E. Edgecombe. A simple measure of electron localization n atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397-5403.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, J. Cohen, A. W. Yang. Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498-6506.
- E.R. Scerri. Have Orbitals Really Been Observed. J. Chem. Educ. 2000, 77, 1492–1494.

