 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anup Pandith | -- | 6141 | 2022-08-24 08:57:22 | | | |
| 2 | Lindsay Dong | + 362 word(s) | 6503 | 2022-08-29 07:30:40 | | | | |
| 3 | Lindsay Dong | -3 word(s) | 6500 | 2022-08-29 07:46:27 | | | | |
| 4 | Lindsay Dong | -1 word(s) | 6499 | 2022-08-29 07:49:53 | | | | |
| 5 | MADHUSUDAN DASNUR NANJAPPA | + 522 word(s) | 7021 | 2022-09-03 20:00:14 | | |
Video Upload Options
Alkaline phosphatase (ALP) is one of the vital phospho-ester bond cleaving biocatalysts that has inevitable significance in cellular systems, viz., early-stage osteoblast differentiation, cell integrity in tissues, bone mineralization, cancer biomarker, liver dysfunction, cellular osmotic pressure, protein folding and many more. Variation from optimal levels of ALP in intra and extracellular fluids can cause severe diseases, including death. Due to these reasons, ALP is considered as a vital biomarker for various preclinical and medical diagnosis. Fluorescence image-based diagnosis is the most widely used method, owing to its simplicity, robustness, non-invasive properties and excellent spatio-temporal resolution (up to the nM/pM level), as compared to conventional analytical techniques, such as the electroanalytical method, nuclear magnetic resonance (NMR) and high-performance liquid chromatography (HPLC). Most of the reviews reported for ALP’s recognition in the literature scarcely explain the structurally related, photophysical and biophysical parameters; and the sub-cellular localizations. Considering these facts, in order to enhance the opto-analytical parameters of fluorescence-based diagnostic materials at the cellular level, herein we have systematically documented recent developments in the opto-analytical capabilities of quencher-free probes for ALP, used in in vitro (biological buffers) to in cellulo conditions, along with in vivo models.
1. Introduction
2. Conceptual Strategies for the Design of ALP Fluorescent Probes in Cellulo Recognition
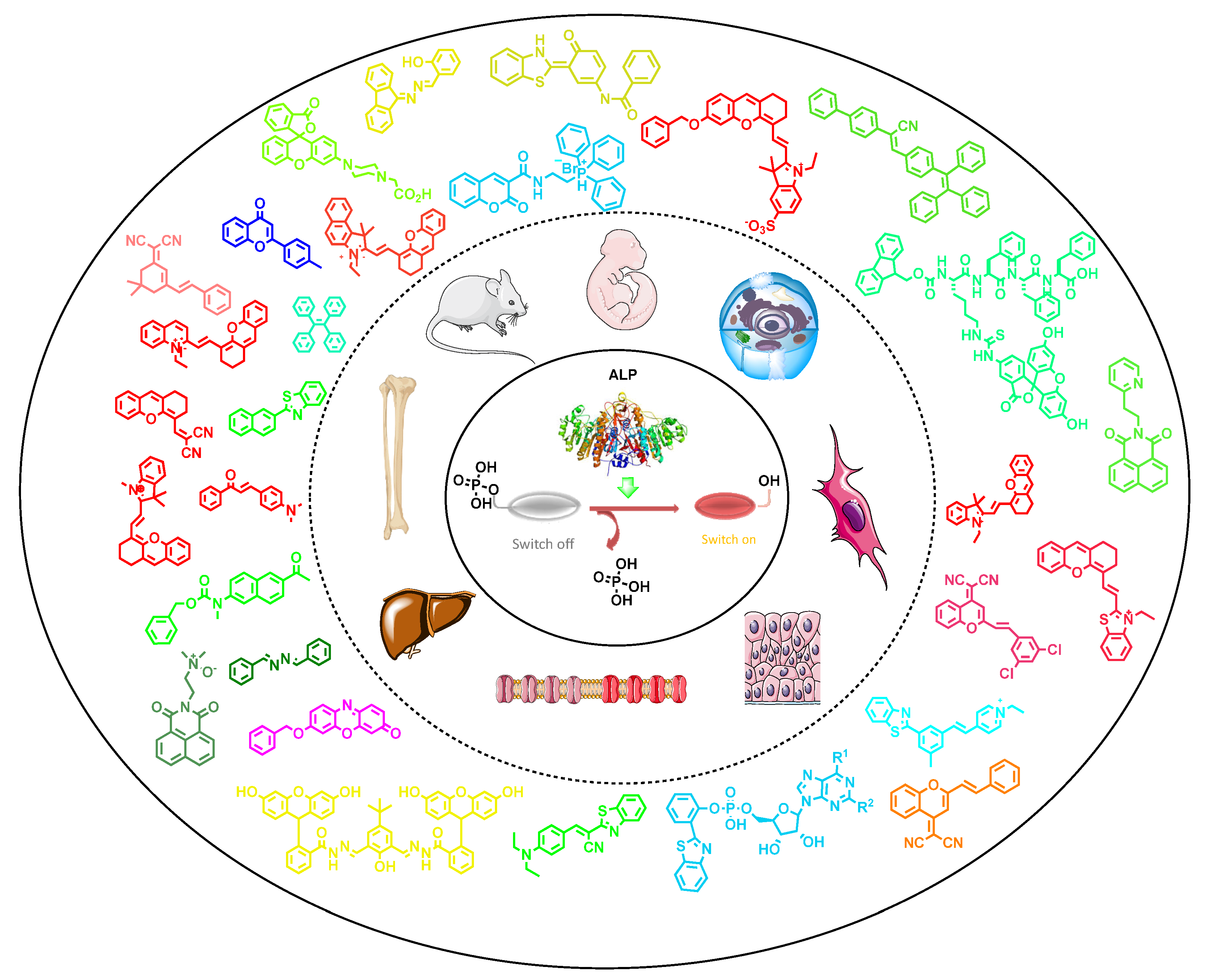
3. Small-Molecule-Based Fluorescent Probes
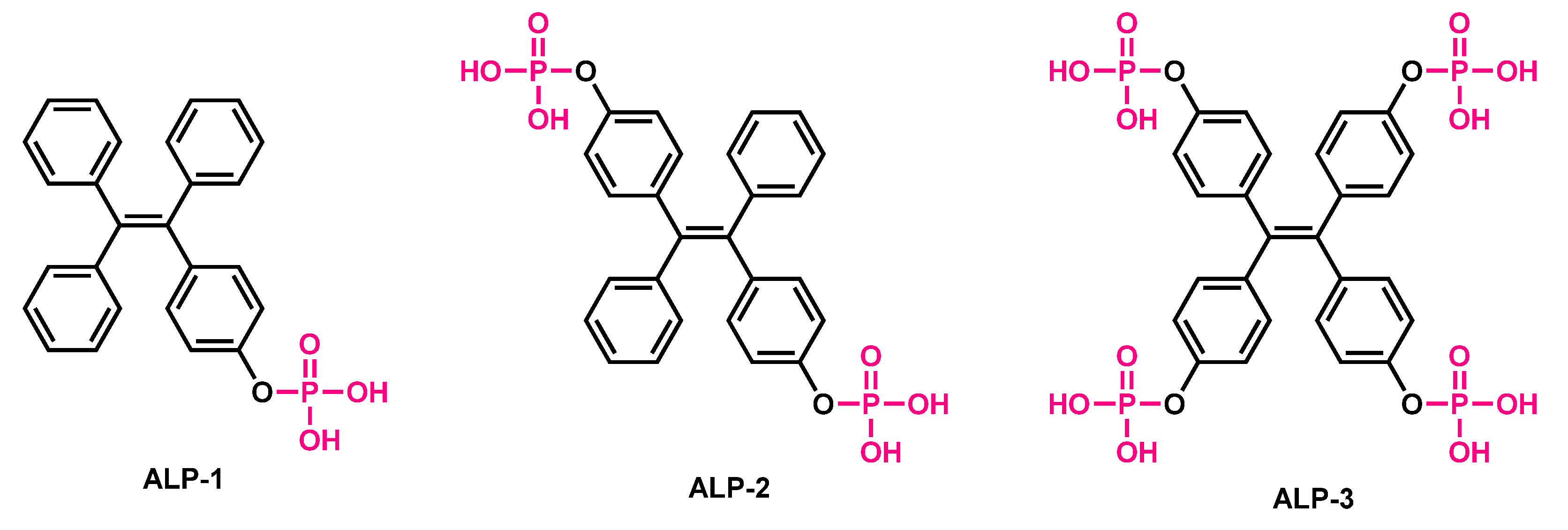
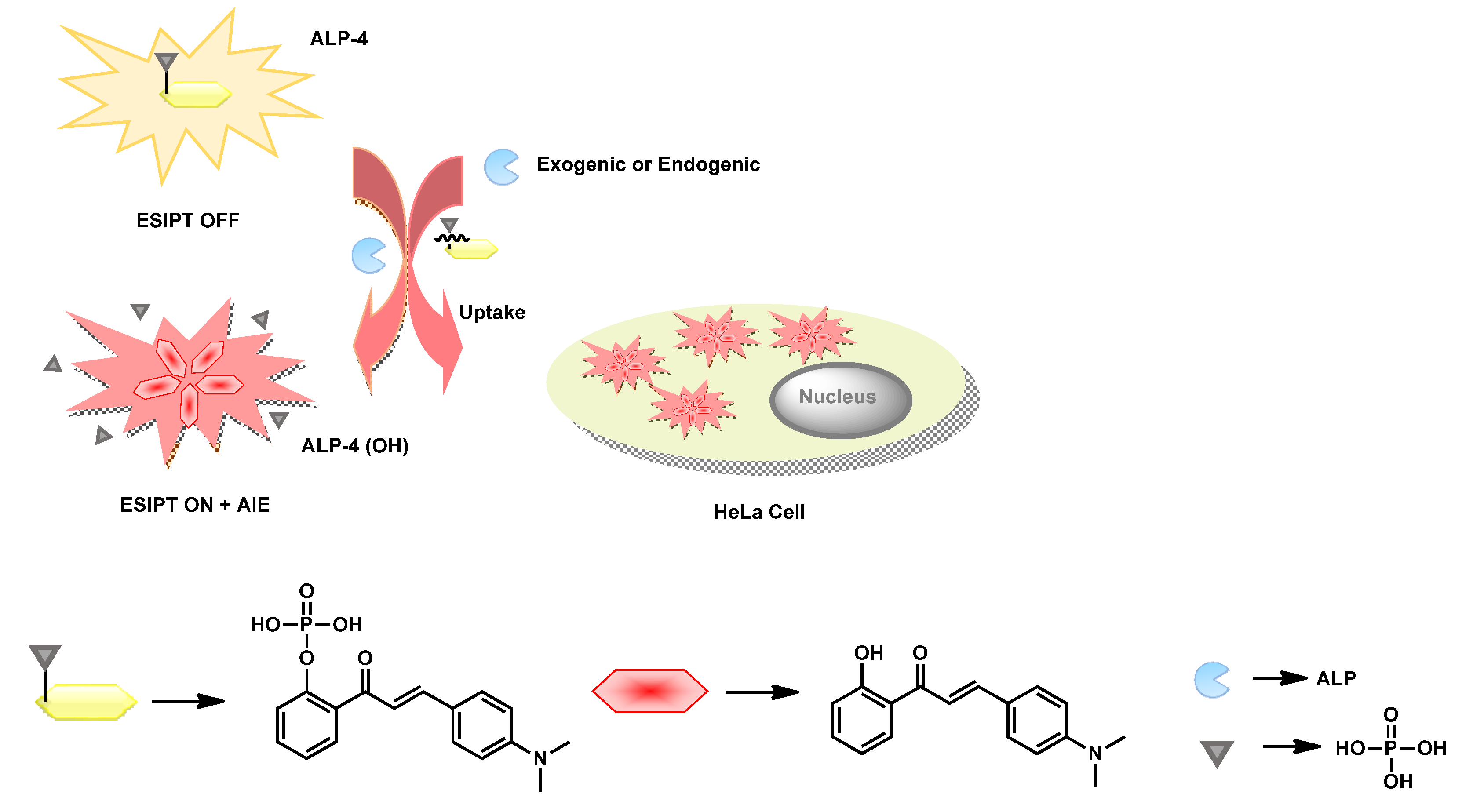
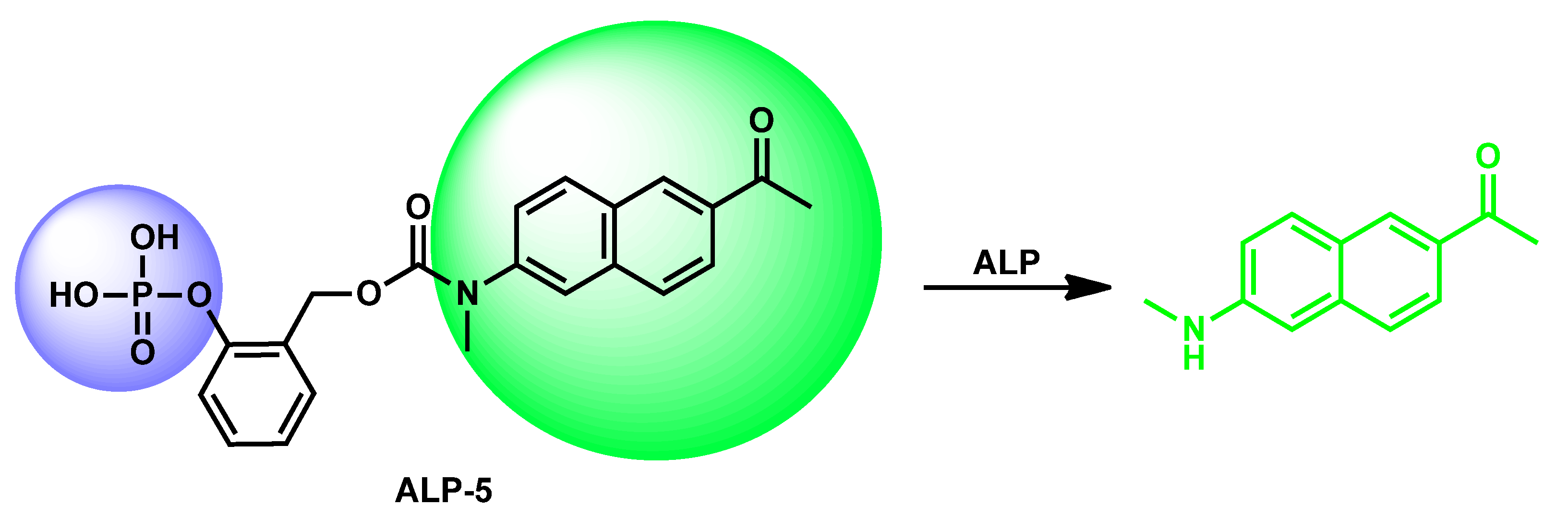
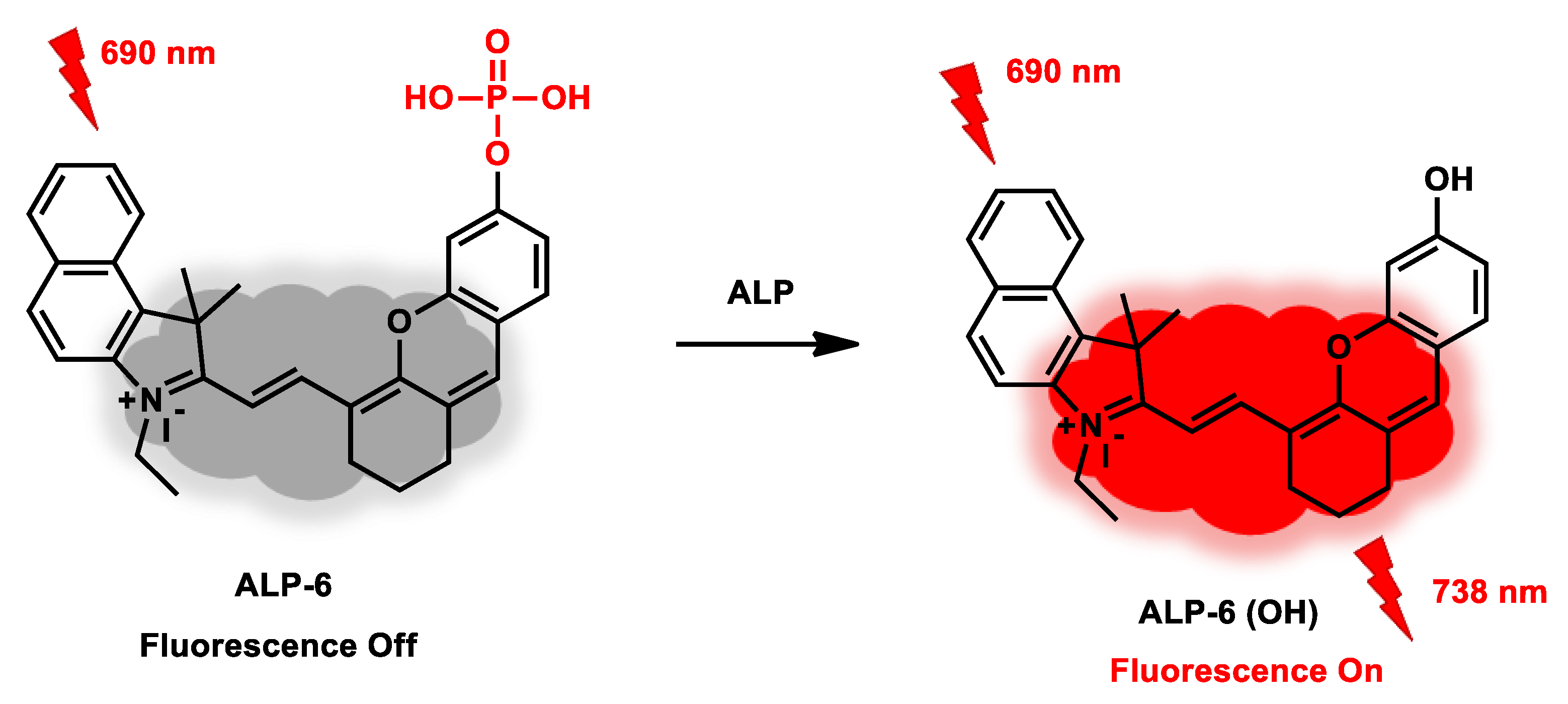
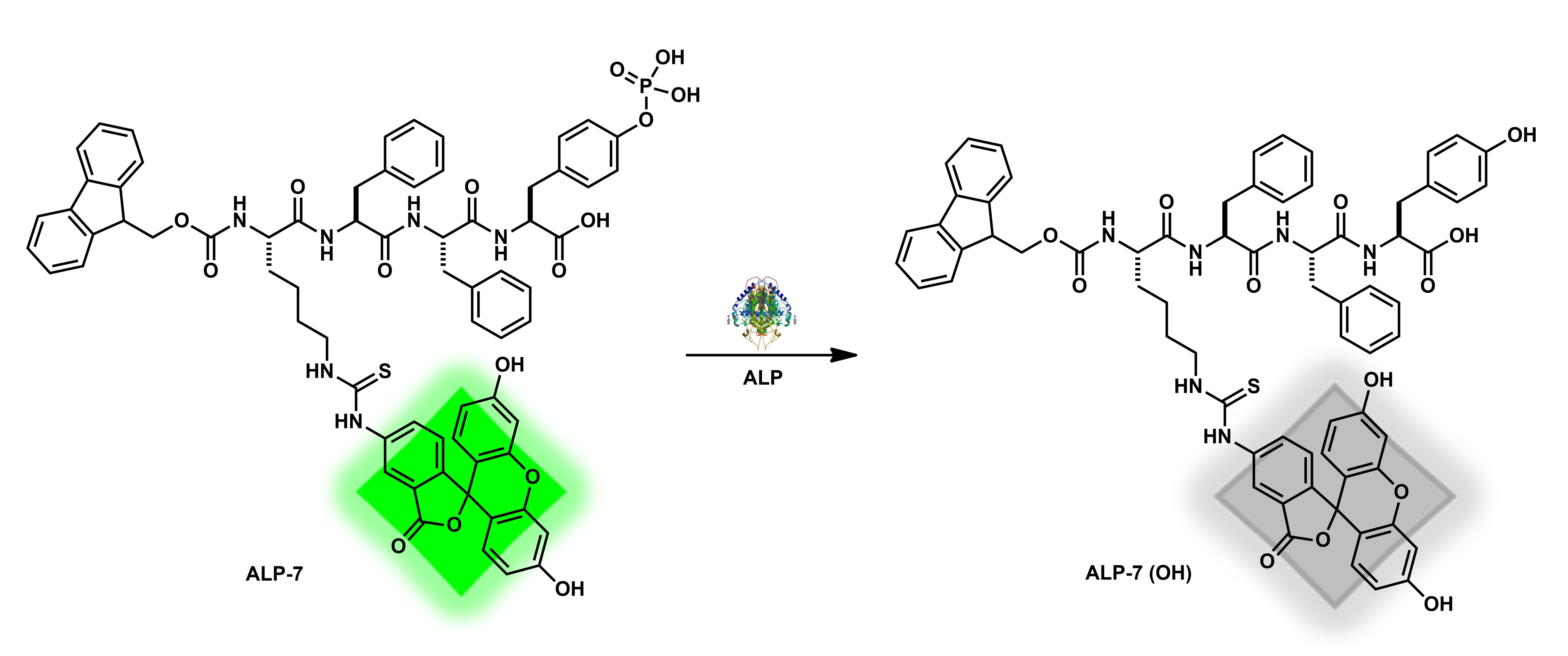
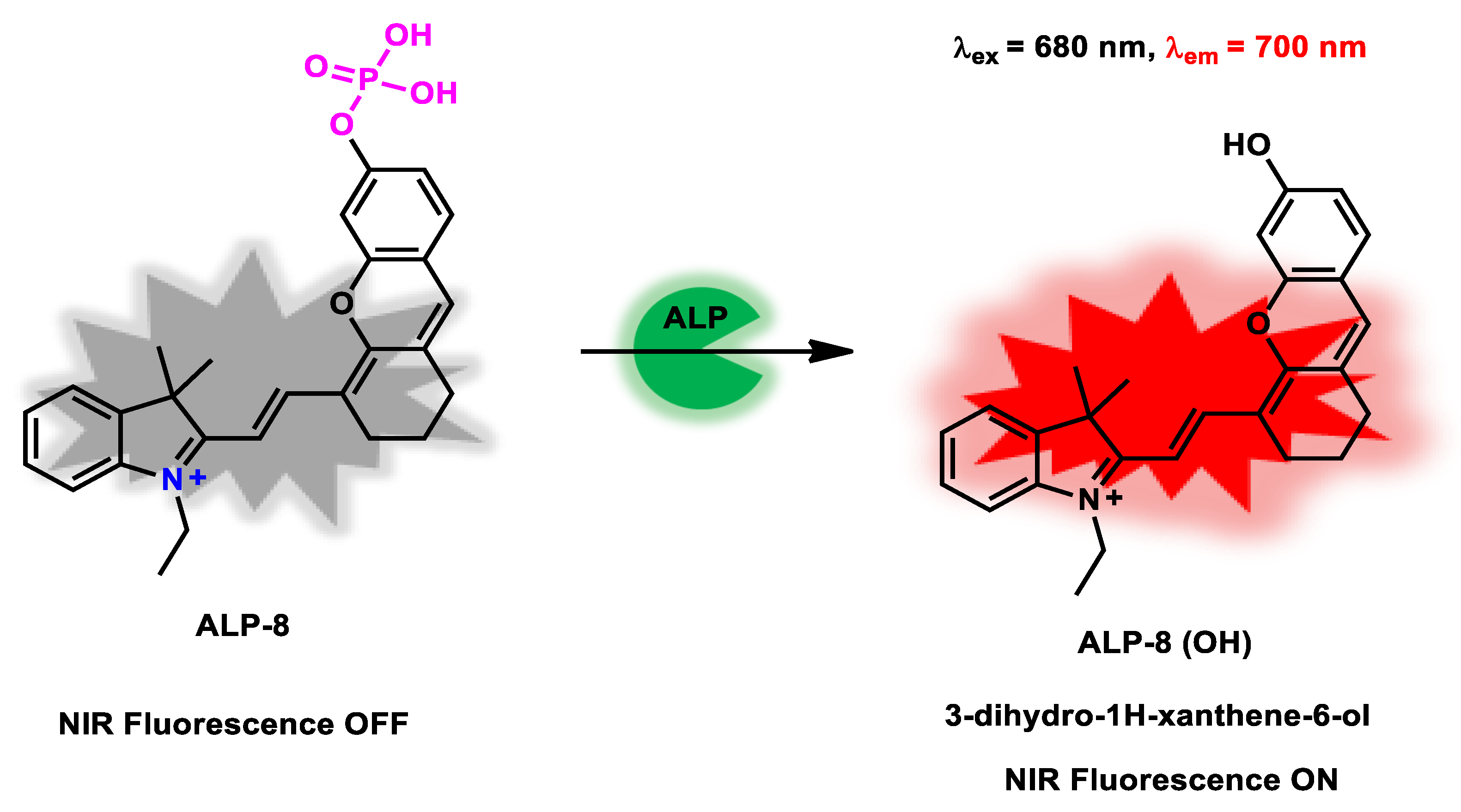
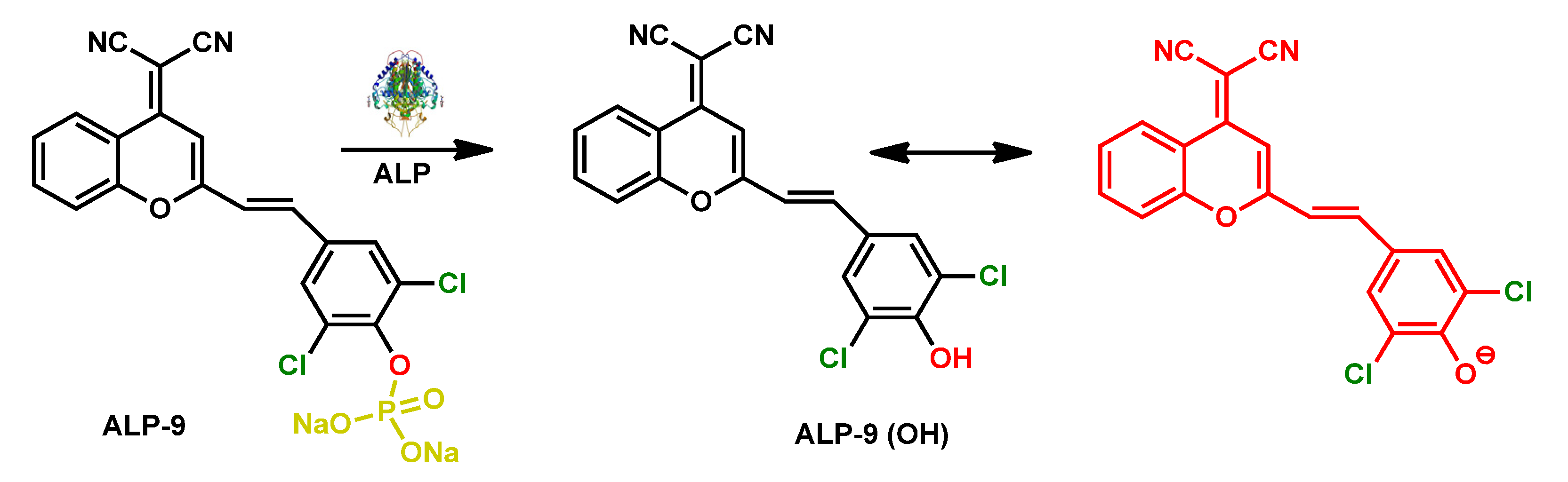
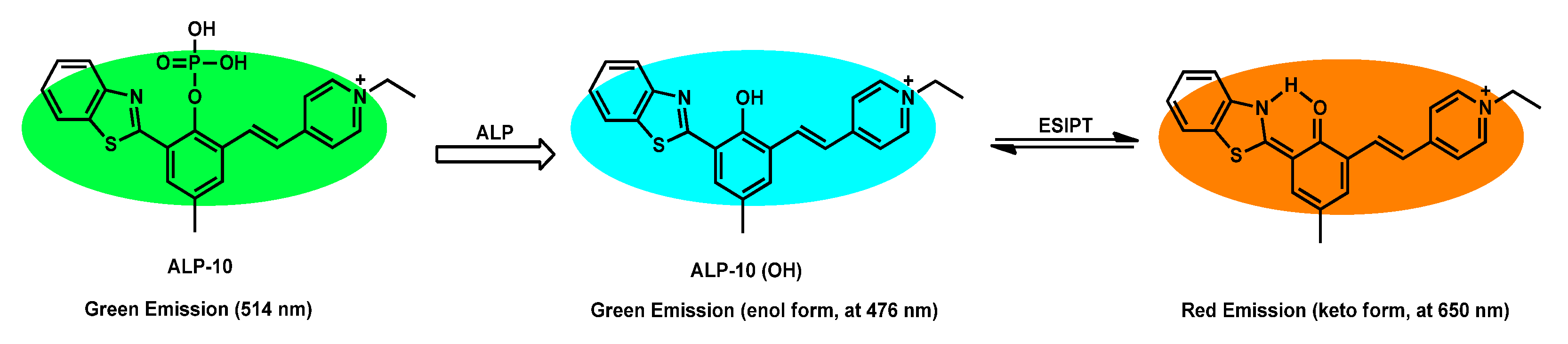
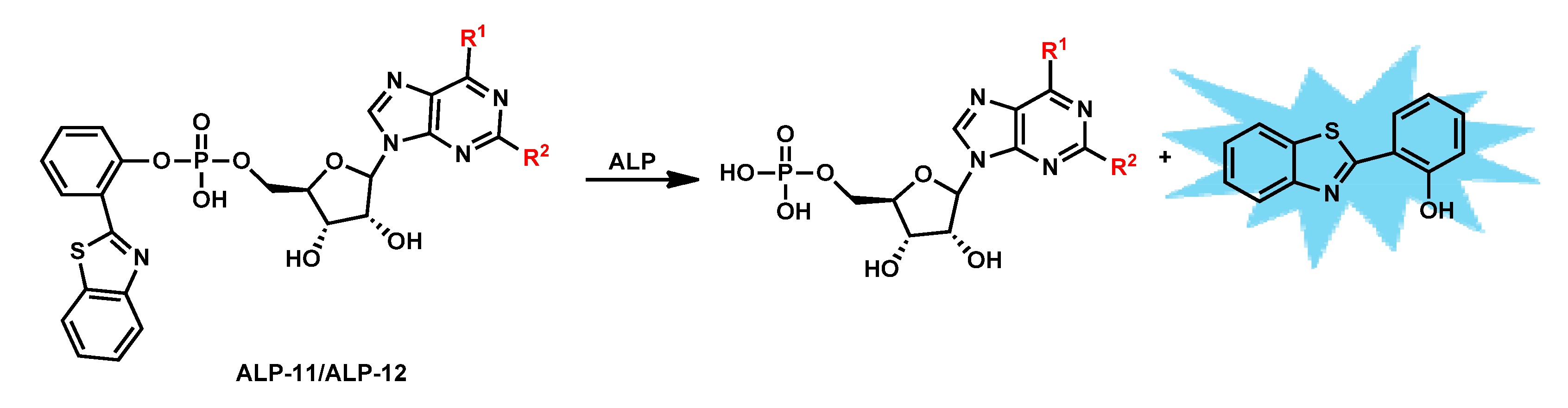
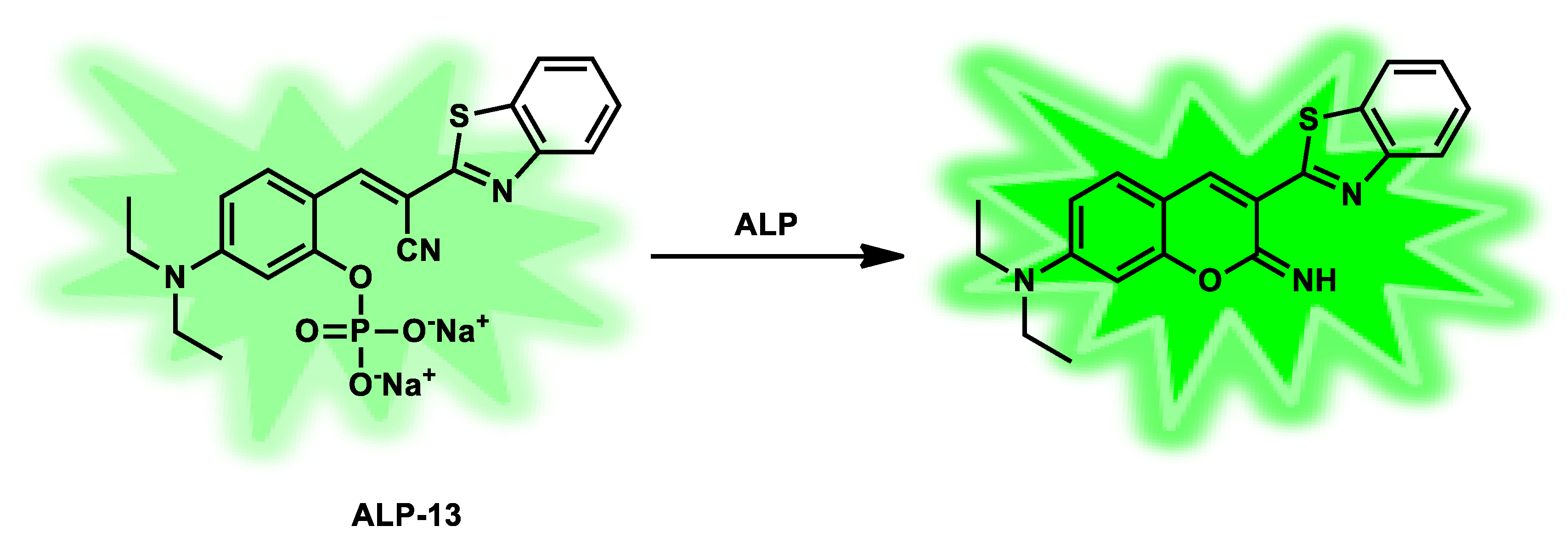
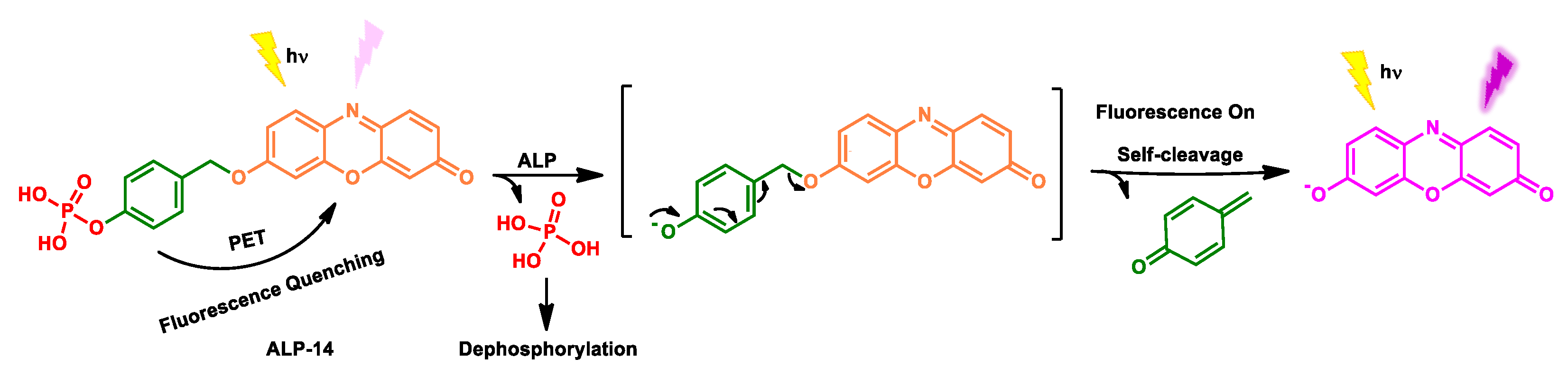
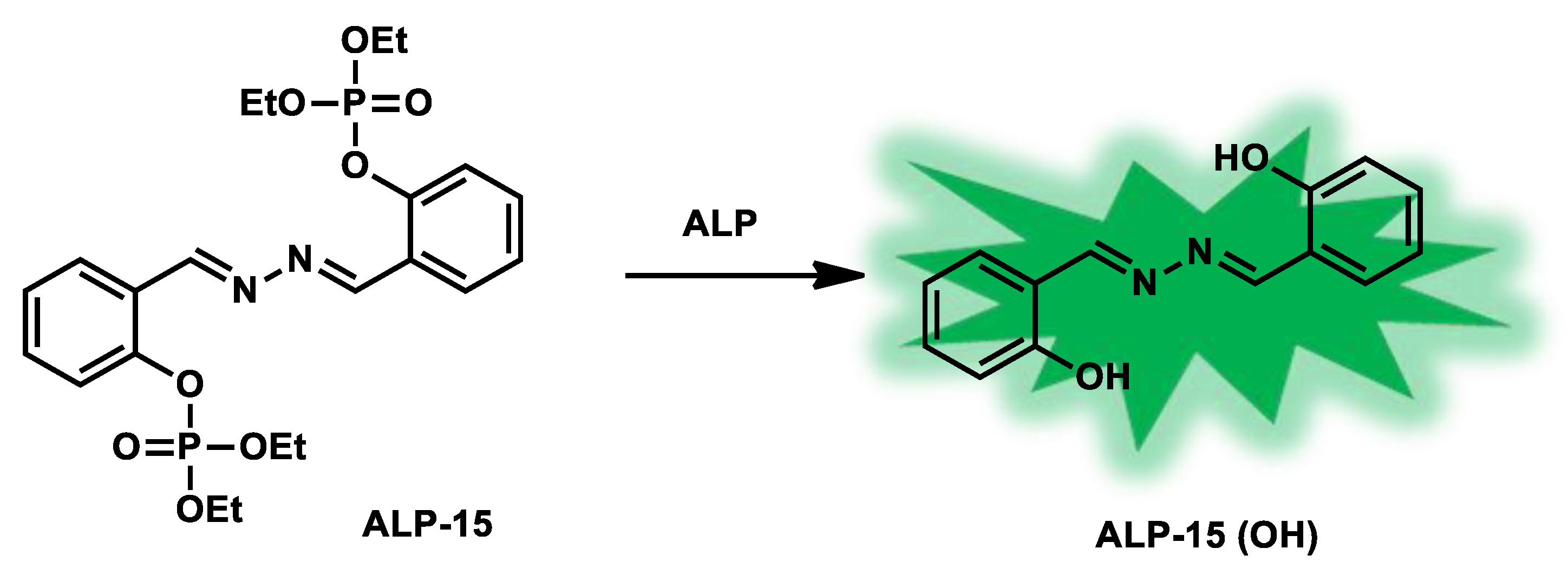
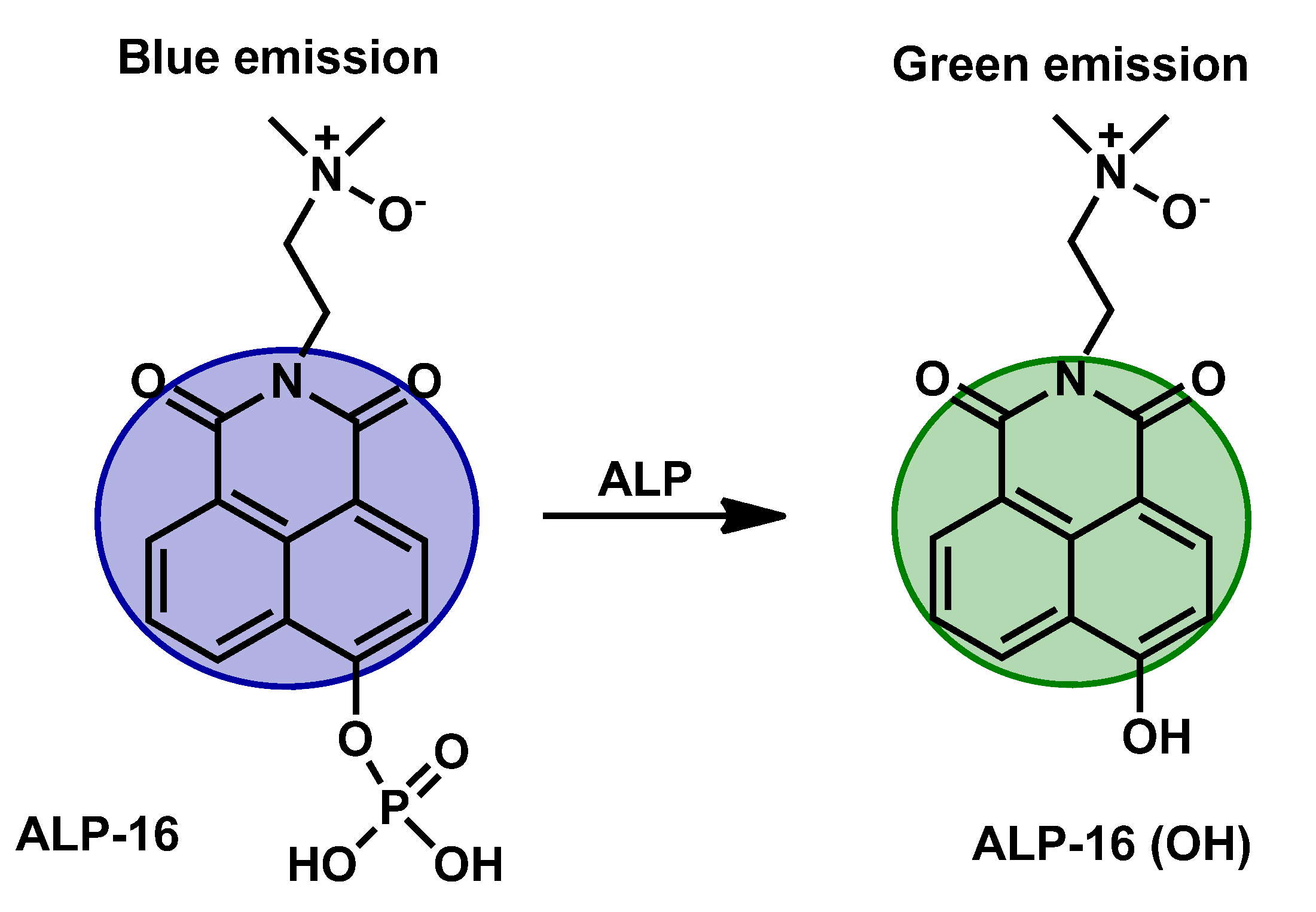
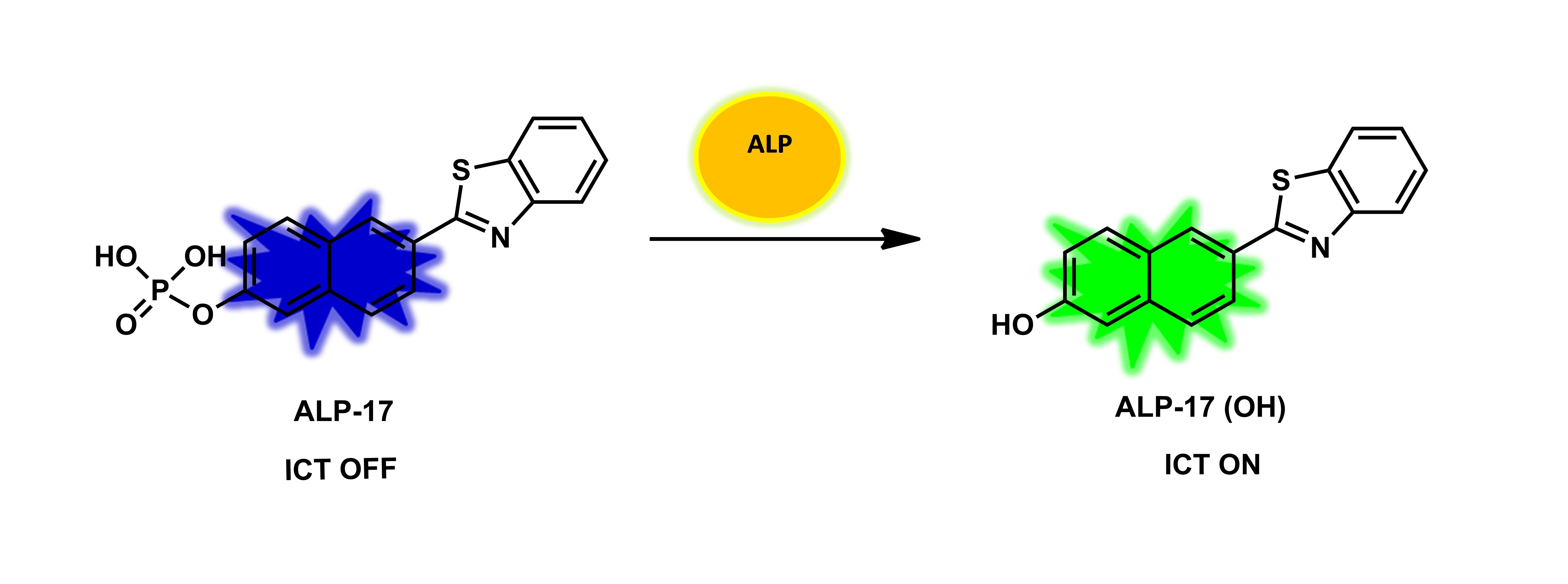 Figure 15. The structure of ALP-17 and a plausible phosphatase recognition mechanism.
Figure 15. The structure of ALP-17 and a plausible phosphatase recognition mechanism.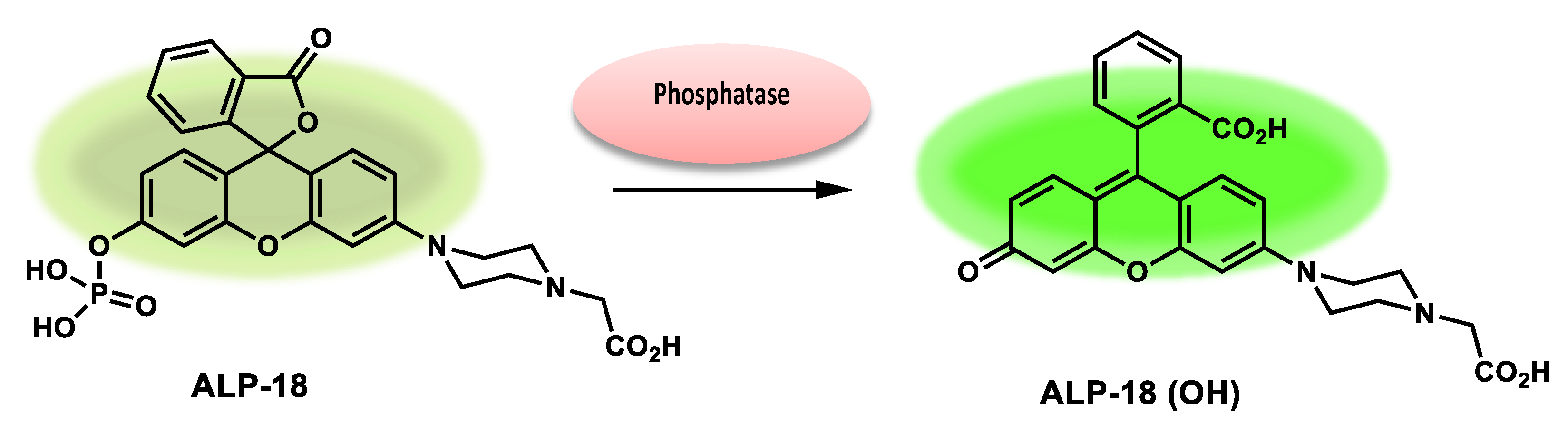
| Probes | Linear Range | kcat/KM Value | Detection Limit |
Response Time |
Stokes Shift |
λex/λem (nm) | Quantum Yield (Φ) |
IC50 Value | Sensing Mode | Application | Cytotoxicity | Cellular Localization |
Ref. |
| ALP-1, ALP-2, ALP-3 |
10–50 mU/mL, 10–40 mU/mL |
/ | / | / | / | 338/469, 337/457, 337/469 |
0.075, 0.118, 0.101 |
/ | Turn-on Fluorescence |
Cellular imaging in living stem cells |
30 µM | Bone marrow mesenchymal stem cells |
[71] |
| ALP-4 | 0–150 U/L | / | 0.15 U/L | / | 211 nm | 430/539, 641 | / | / | Ratiometric | Live cell imaging | 20 µM | Intercellular fluids | [72] |
| ALP-5 | 0.5–10 U/L | / | 0.30 U/L | / | 135 nm | 365/500 | / | 7.39 µM | Turn-on Fluorescence |
In living cells and tissues |
20 µM | Cellular and tissue lysates, cytoplasm |
[73] |
| ALP-6 | 0.01–2.0 U/mL | / | 0.003 U/mL | 20 min | / | 690/738 | / | 141.9 µM | Turn-on Near-Infrared Fluorescence |
In Vivo Cell, tissue and living animal |
30.0 µM | / | [74] |
| ALP-7 | 0–2.8 U/mL | / | 0.06 U/mL | 2 h | / | 465/530 | / | / | Turn-Off Fluorescence |
in Vitro and in Living Cells |
160 µM | / | [75] |
| ALP-8 | 0–0.1 U/mL | 1.01 × 105 M−1 s −1 |
0.07 U/L | 2 min | / | 680/700 | / | 7.51 µM | Off−On Near-infrared Fluorescence |
Living Cells and Mice |
20 µM | / | [76] |
| ALP-9 | 0.5–8.0 U/L | / | 0.072 U/L | / | / | 535/683 | / | / | Rational Turn-on Red-Near-Infrared fluorescence |
In vivo and in vitro |
50 µM | Mitochondria, Lysosome |
[77] |
| ALP-10 | 0–60 mU/mL | / | 0.072 mU/mL |
/ | 260 nm | 390/476, 650 | 0.16 | 10 µM | Ratiometric | living cells and in vivo |
10 µM | Mitochondria | [78] |
| ALP-11, ALP-12 | / | / | / | / | / | 405/500–530 | / | 10 µM | Turn-on Fluorescence |
Living cells and zebrafish model |
10 µM | / | [79] |
| ALP-13 | / | 1.4 × 104 M−1 s −1 |
/ | / | / | 472/542 | 0.002 | 36 µM | Turn-on Fluorescence |
Living cells | 10 µM | / | [80] |
| ALP-14 | / | 1.7 × 104 M−1 s −1 |
1.09 U/L | / | / | 550/585 | 0.0023 | 7.58 µM | Turn-on Fluorescence |
Living cells | / | Cytosol | [81] |
| ALP-15 | 0–10 U/L | / | 0.012 U/L | / | 180 nm | 356/536 | / | / | Turn-on Fluorescence |
Living cells | / | / | [82] |
| ALP-16 | / | / | 0.38 U/L | / | / | 425/554 | / | / | Ratiometric | In vivo | 5 µM | / | [83] |
| ALP-17 | 20–180 U/L | / | 2.3 U/L | / | / | 405/410–460, 470–530 |
0.41 | / | Ratiometric | Living cells | 20 µM | / | [84] |
| ALP-18 | / | 13.6 × 105 M−1 s −1 |
0.011 U/L | / | / | 490/540 | 0.0487 | / | Turn-on Fluorescence |
In vitro, cell imaging and cell cultures |
100.0 µM | Lysosome | [85] |
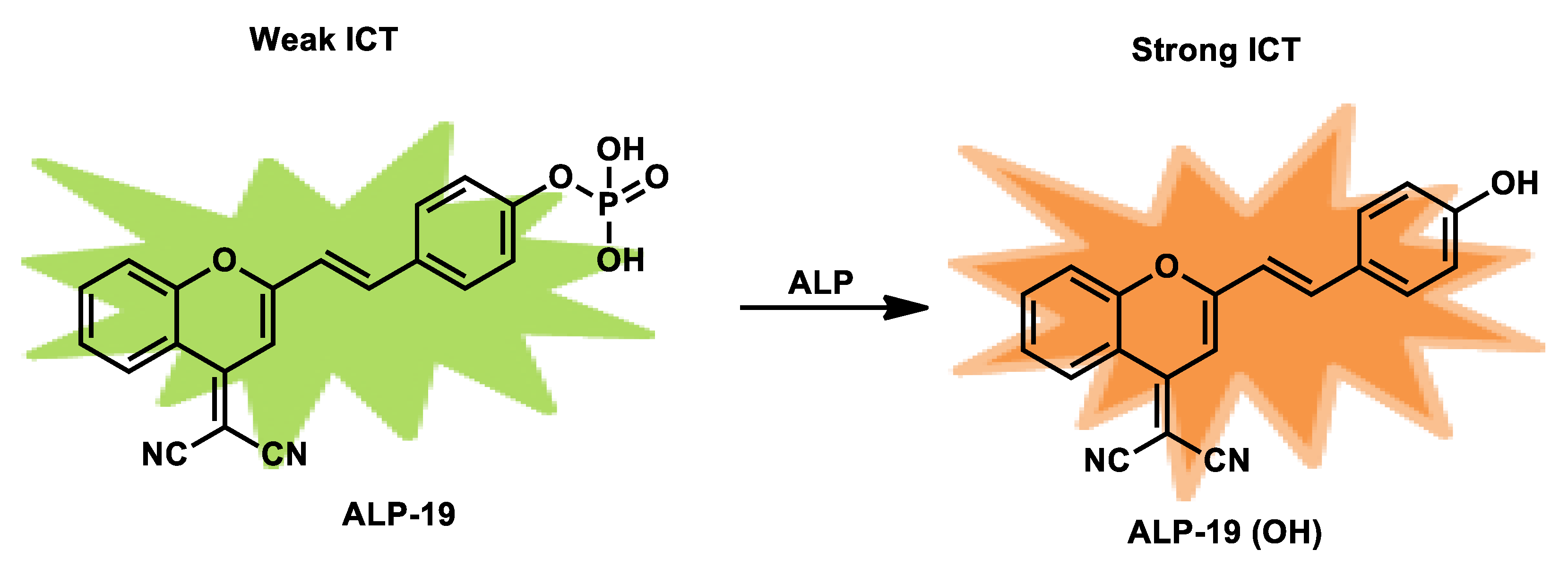
| ALP-19 | 50–200 U/L | / | 3.8 U/L | 30 min | / | 440/550 | 0.105 | / | Ratiometric | Living cells | 10 µM | / | [86] |
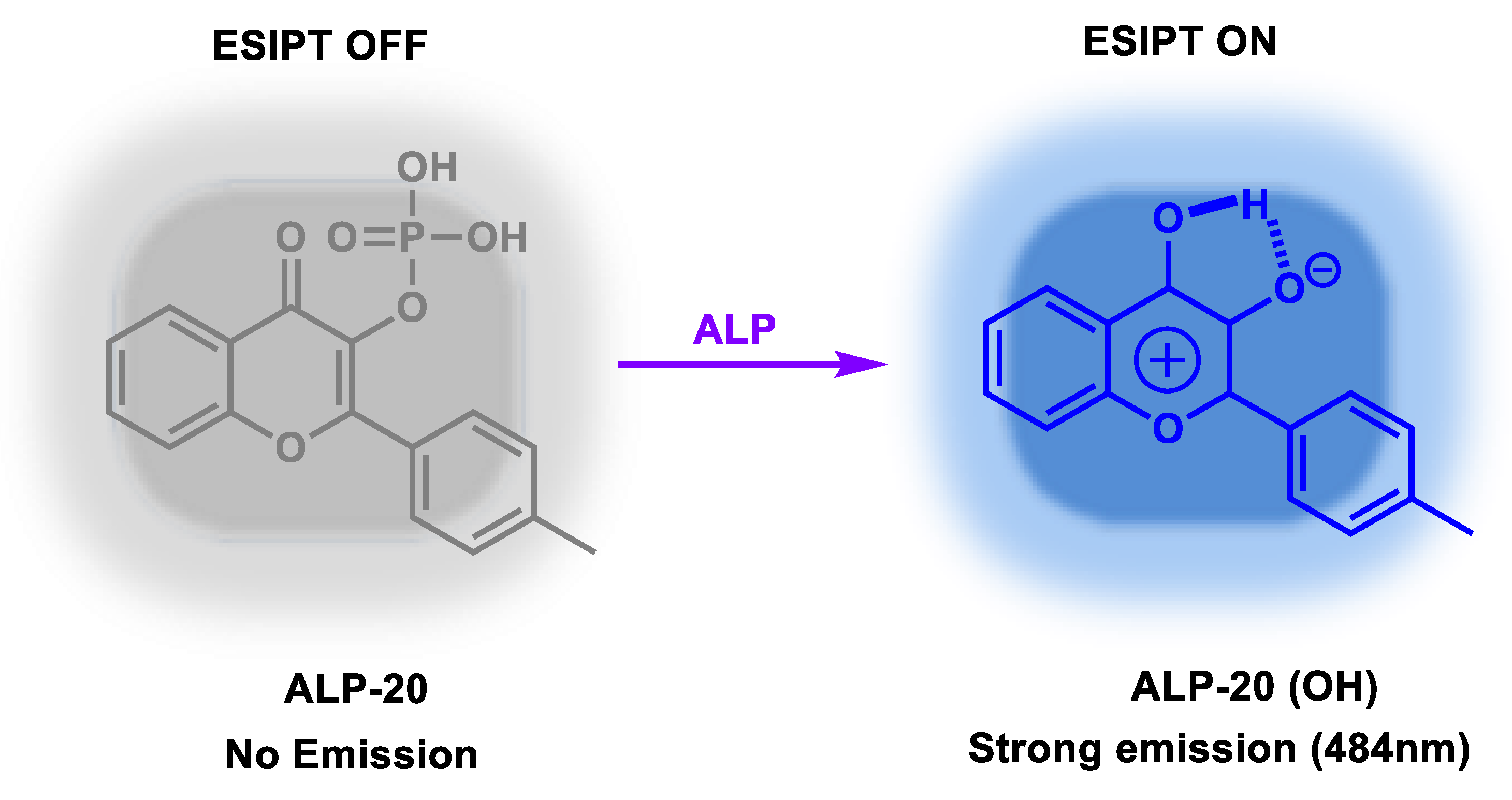
| ALP-20 | / | / | 0.032 U/L | / | / | 400–410/455 | 0.085 | / | Turn-on Fluorescence |
Live cells and serum samples |
50 µM | / | [87] |
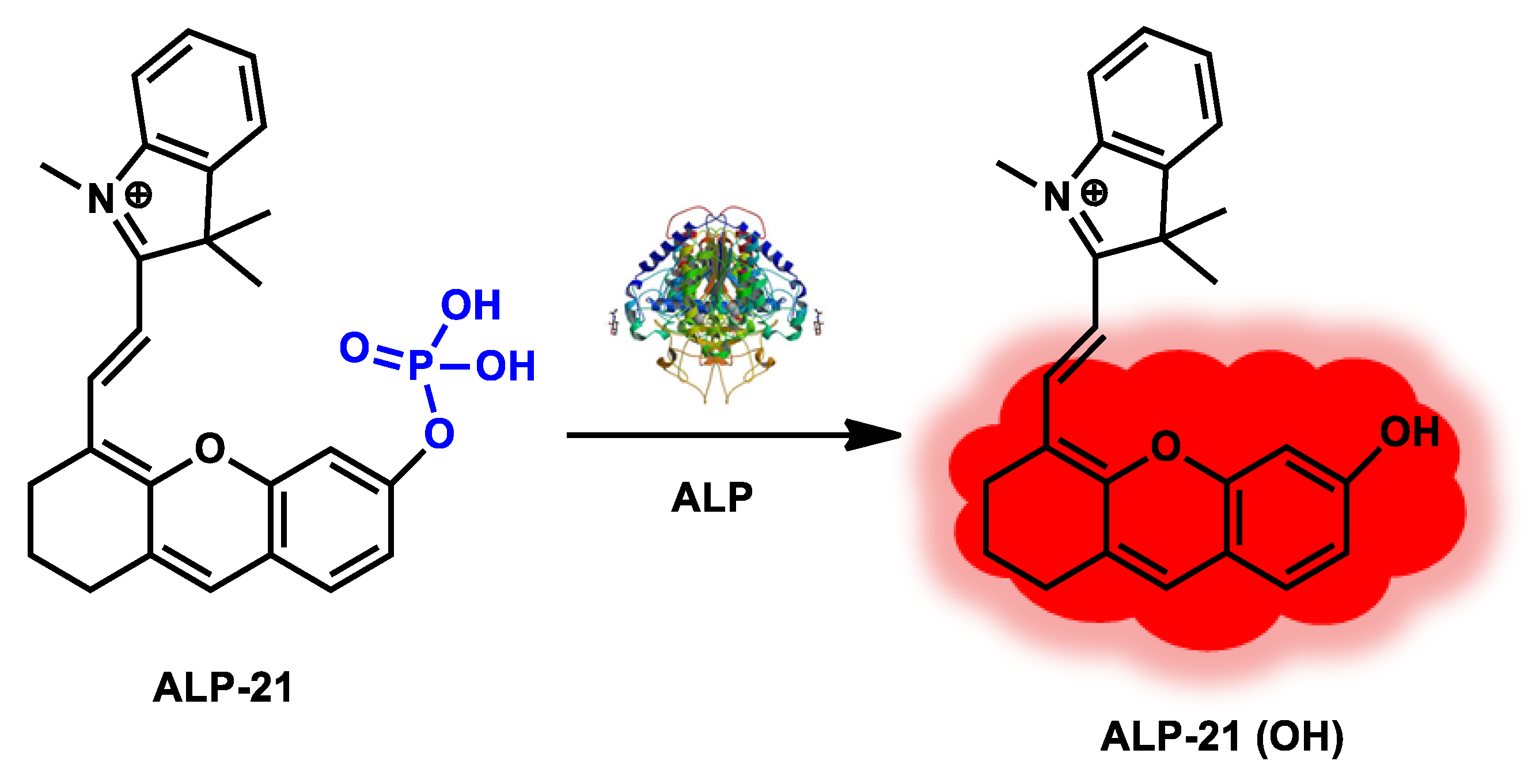
| ALP-21 | 1–30 U/L | / | 0.28 U/L | / | / | 680/690–800 | / | / | Turn-on Fluorescence |
In vivo and in vitro |
20 µM | / | [88] |
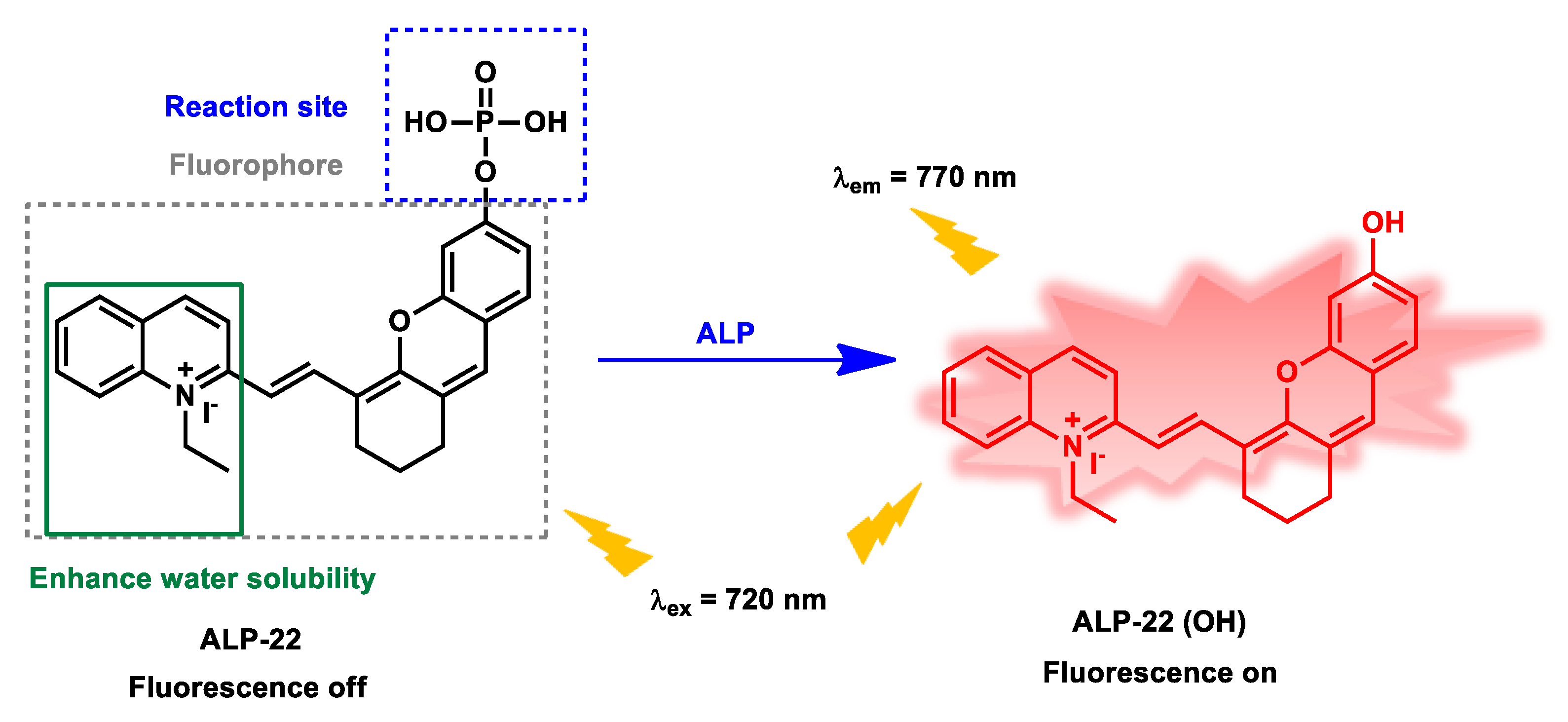
| ALP-22 | 0.05–1.0 U mL−1 |
/ | 0.017 U mL−1 | / | / | 720/770 | / | 109.6 µM | Turn-on Fluorescence |
Cell imaging, treatment of diabetes |
30 µM | / | [89] |
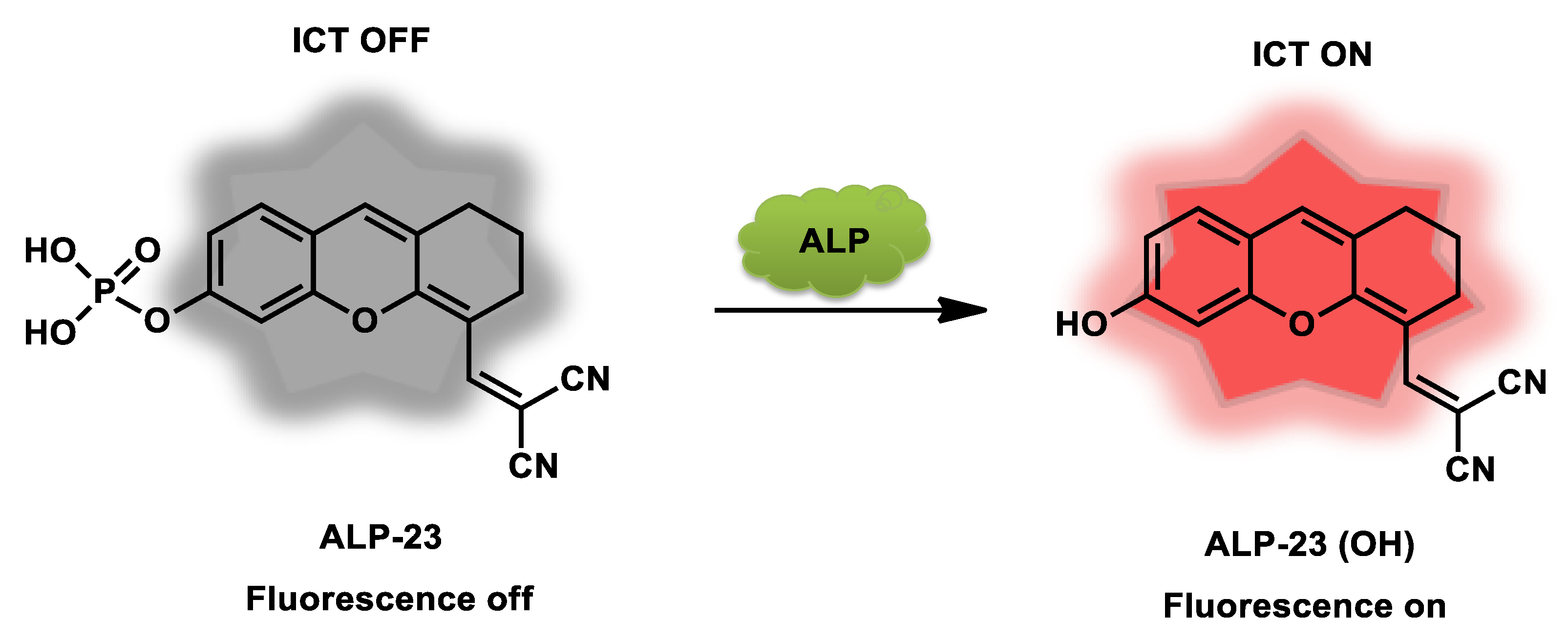
| ALP-23 | 5–100 U/L | / | 0.28 U/L | / | / | 600/620–850 | / | 141.4 µM | Turn-on Fluorescence |
Living cells and zebrafish |
10 µM | / | [90] |
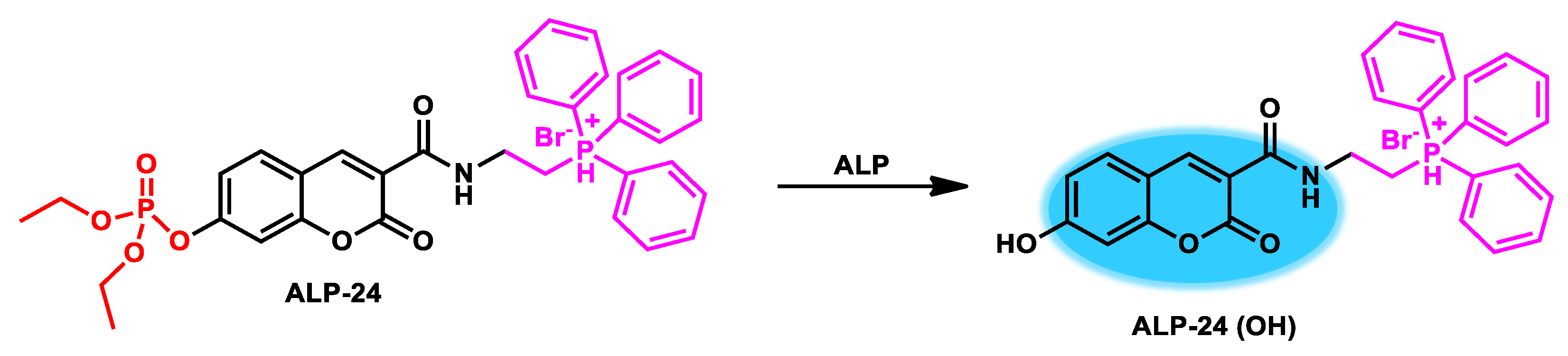
| ALP-24 | / | 3.12 × 106 M−1 s −1 |
2.921 ng/mL | / | / | 410/450 | 0.567 | / | Ratiometric | Live cells | 50 µM | Mitochondria | [91] |
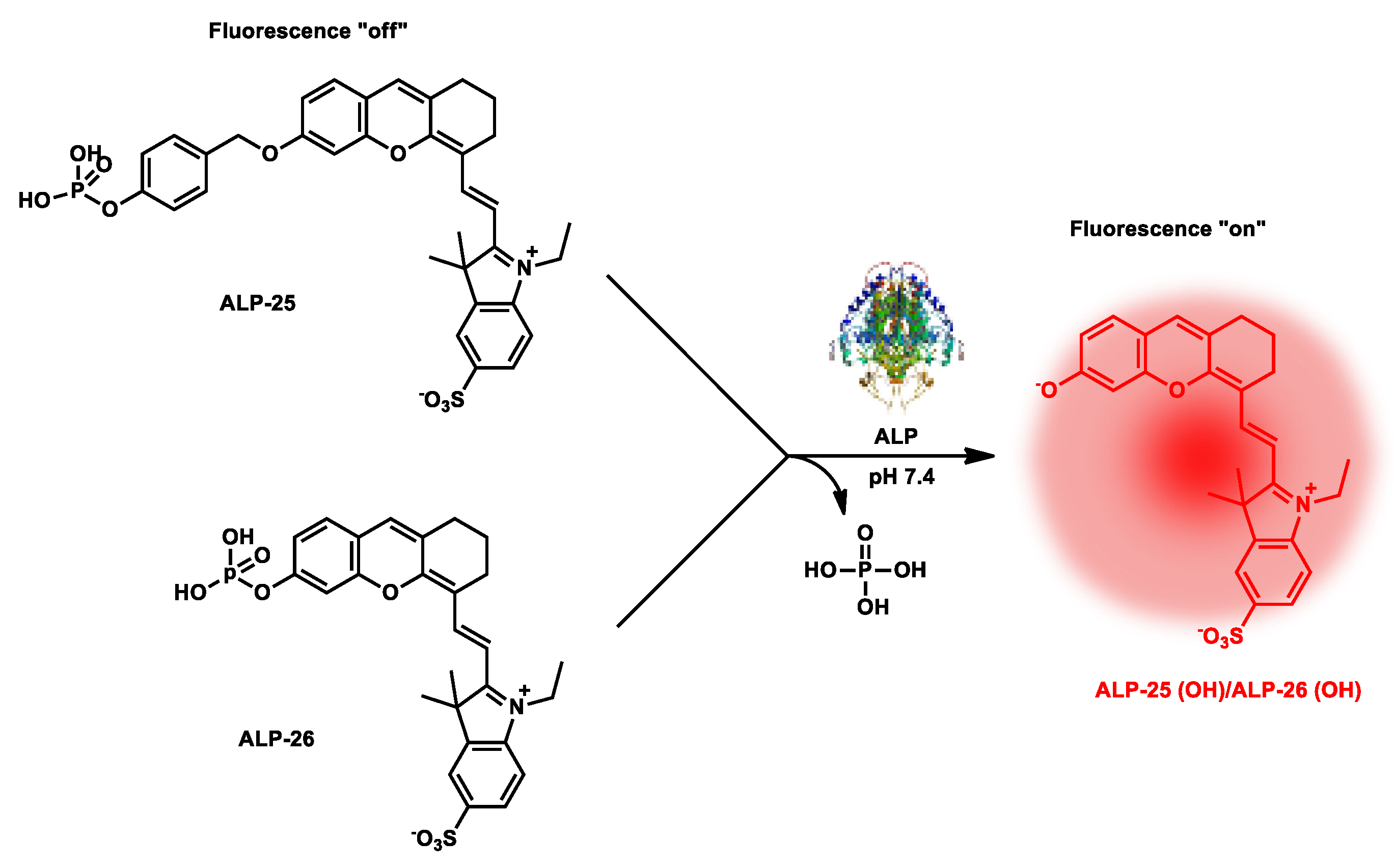
| ALP-25, ALP-26 |
0–1.0 U mL−1 | / | 10−5–10−3 U mL−1 |
1.5 min | / | 685/710 | 0.13 | / | Turn-on fluorescence |
In vivo | 50 µM | / | [92] |
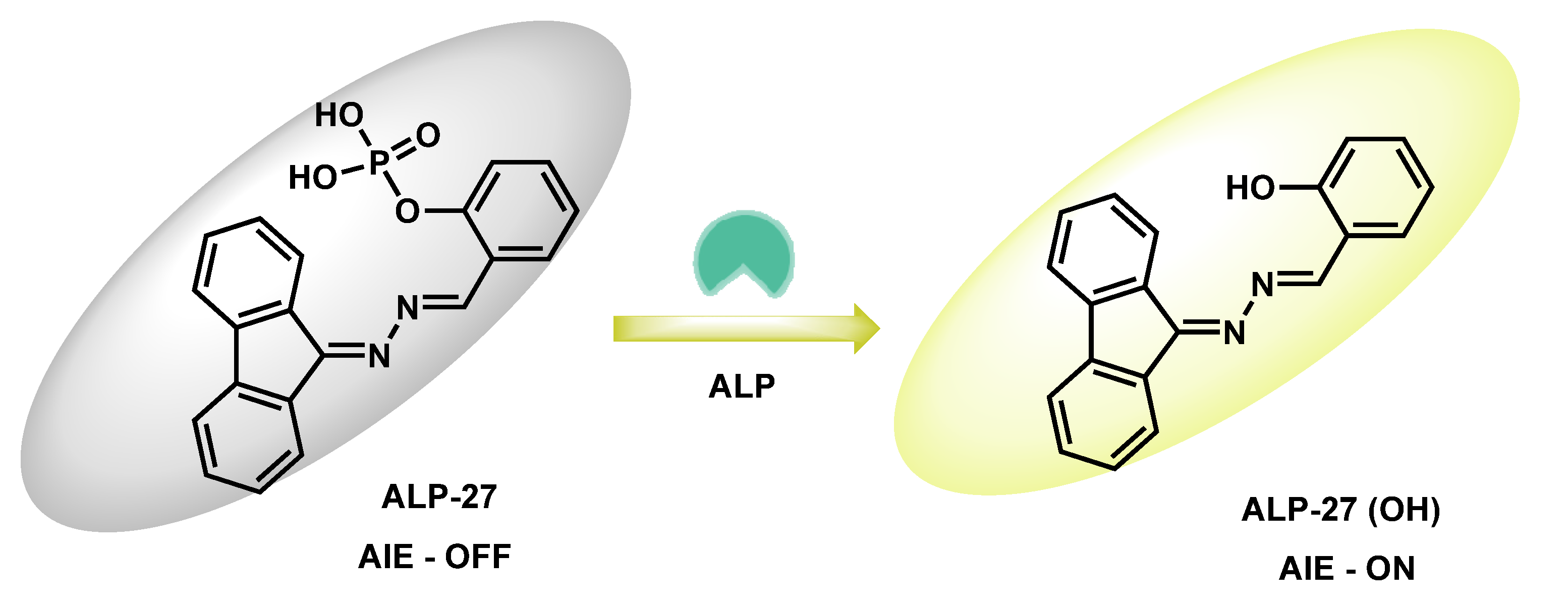
| ALP-27 | 1–100 U/L | / | 0.6 U/L | / | >200 nm | 380/586 | / | / | Turn-on fluorescence |
Living cells | 10 µM | Lipid droplets | [93] |
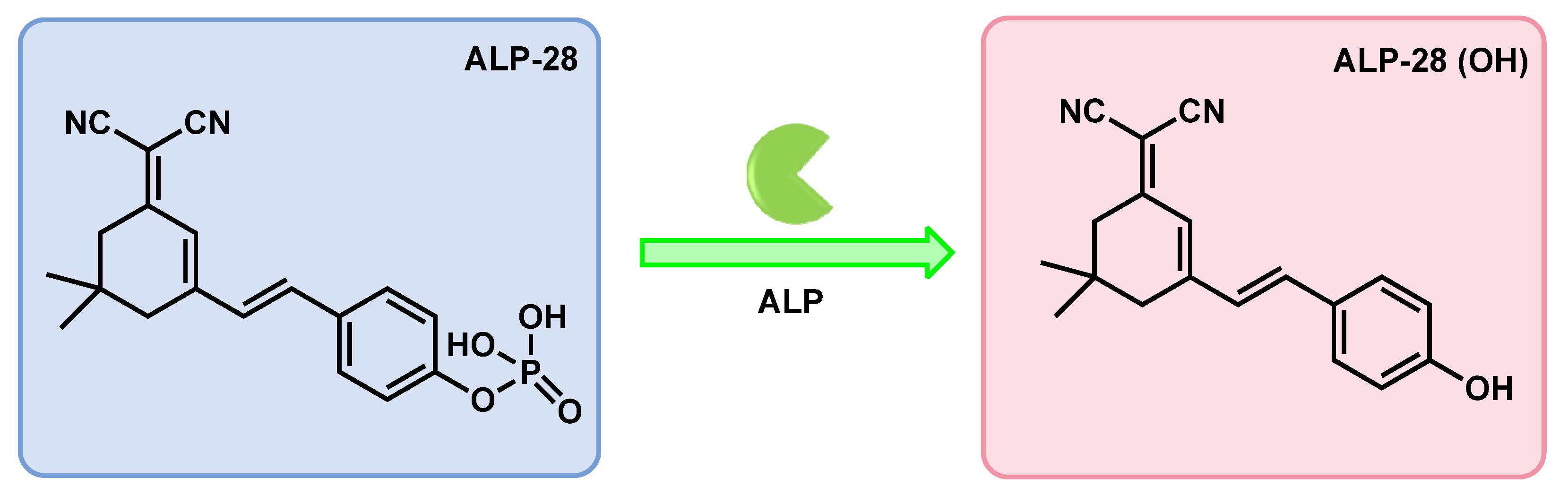
| ALP-28 | / | / | 0.088 U/L | 6 min | / | 440/570 | / | / | Turn-on Fluorescence |
Living cells | 10 µM | / | [94] |
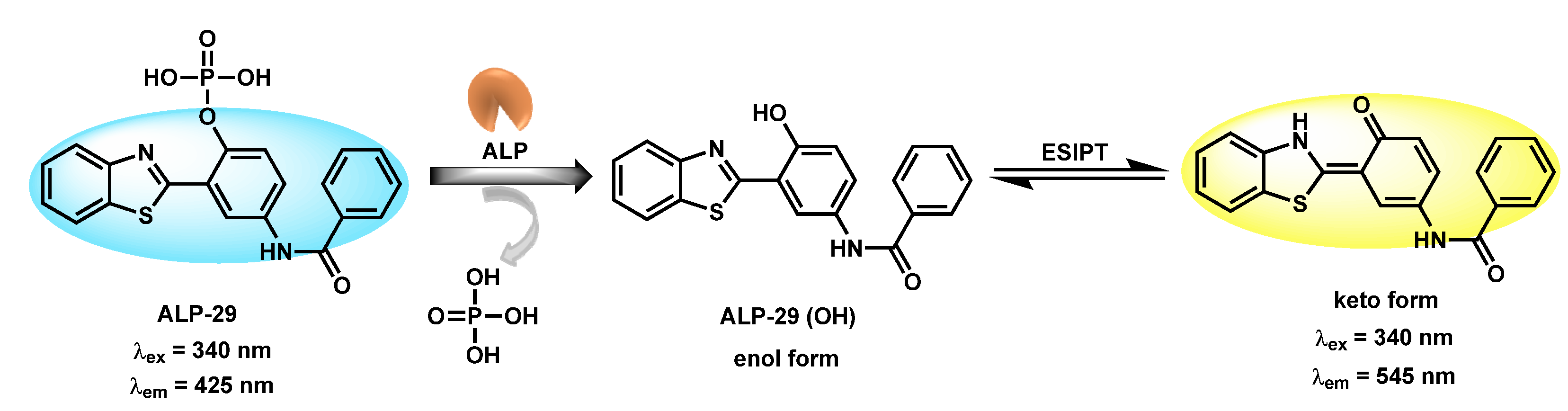
| ALP-29 | / | / | 0.004 mU/mL |
35 min | / | 340/545 | 0.028 | / | Ratiometric | Living cells | 20 µM | / | [95] |
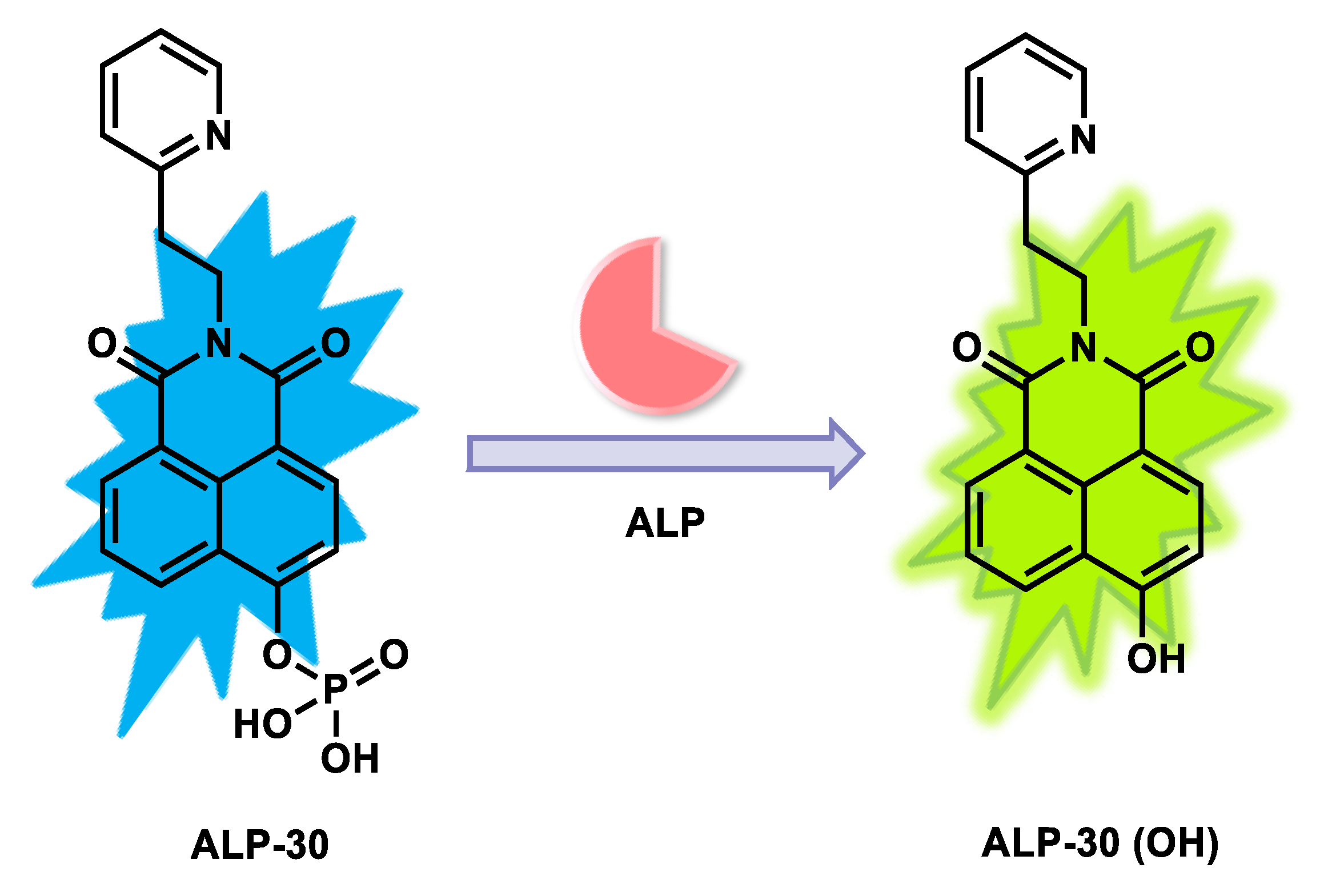
| ALP-30 | 0–200 U/L | / | 0.25 U/L | / | / | 425/472 | / | / | Ratiometric | Living cells | / | / | [96] |
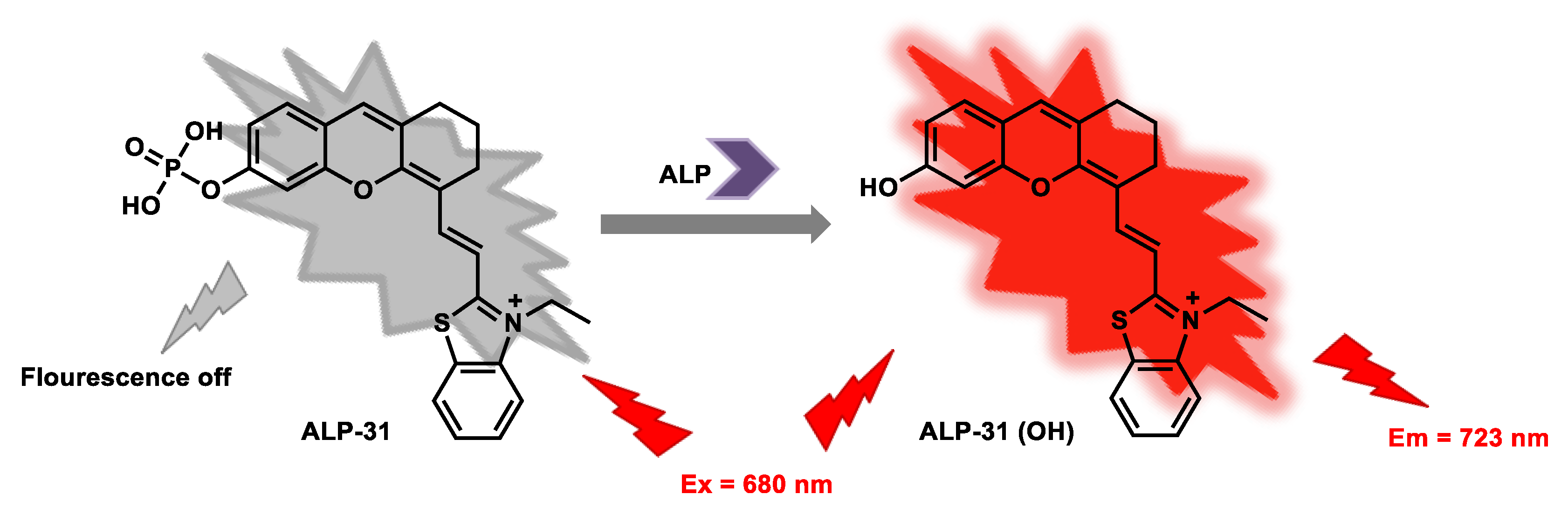
| ALP-31 | 0–8 U/L | / | 0.042 U/L | / | 43 nm | 680/723 | 0.15 | 23.98 µM | Turn-on Near-Infrared Fluorescence |
Living cells | 20 µM | / | [97] |
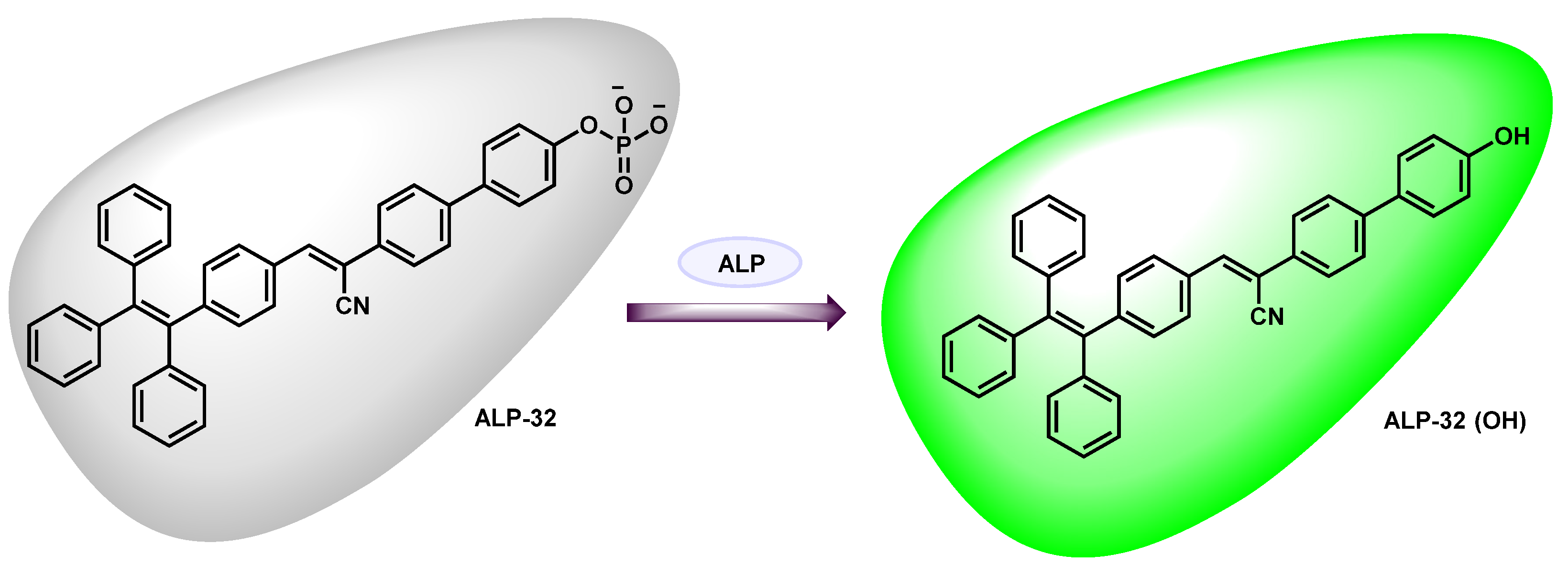
| ALP-32 | / | / | 14.2 U/L | / | / | 440/543 | / | / | Turn-on Fluorescence |
Live cells | 50 µM | / | [98] |
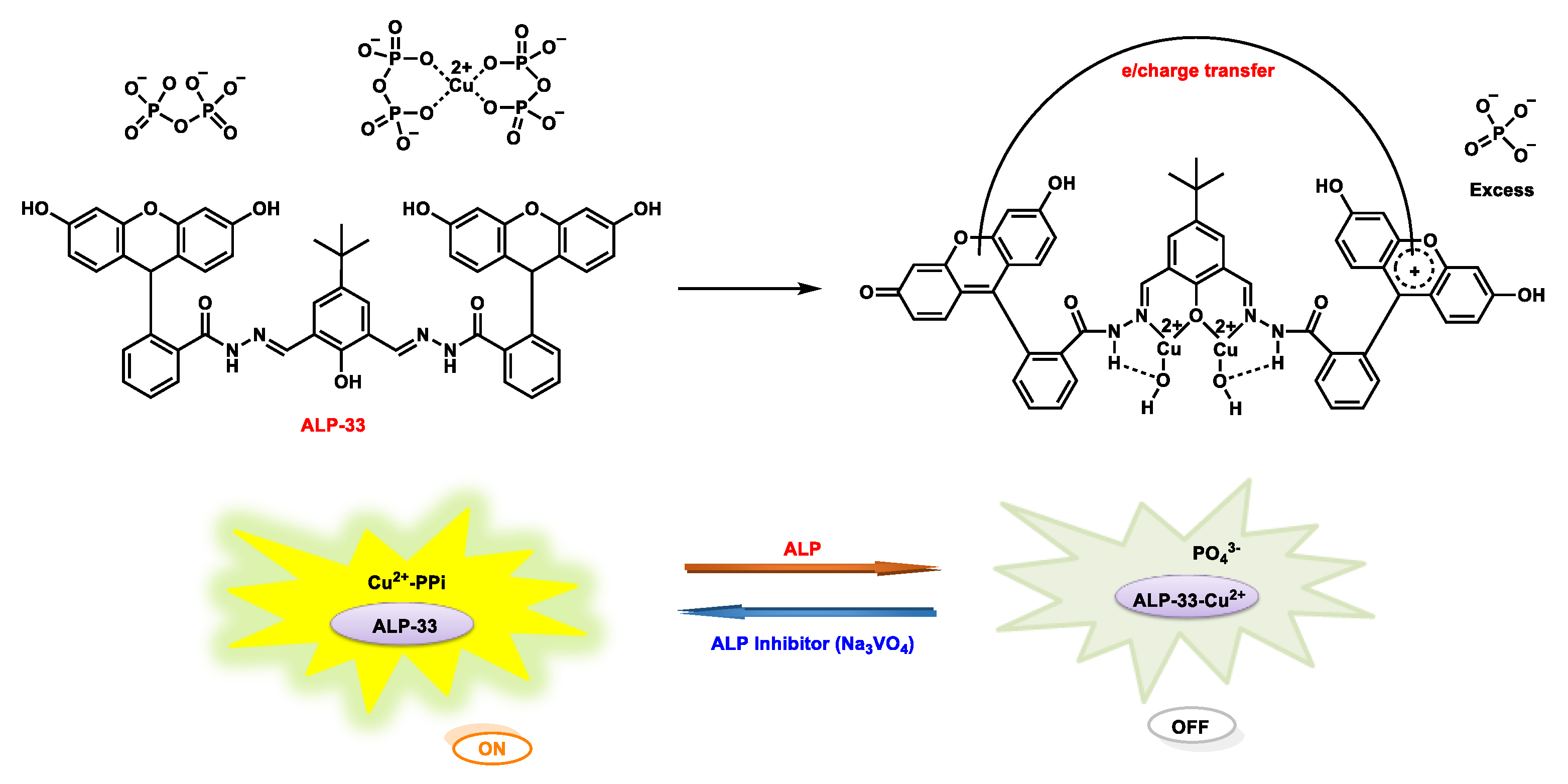
| ALP-33 | 0.05–1.05 U/mL |
/ | 0.012 U/mL | / | / | 377/572 | 0.0392 | / | Colorimetric and fluorescent on-off-on |
Over expressed cancer cells & normal salivary gland cells |
25 µM | / | [99] |
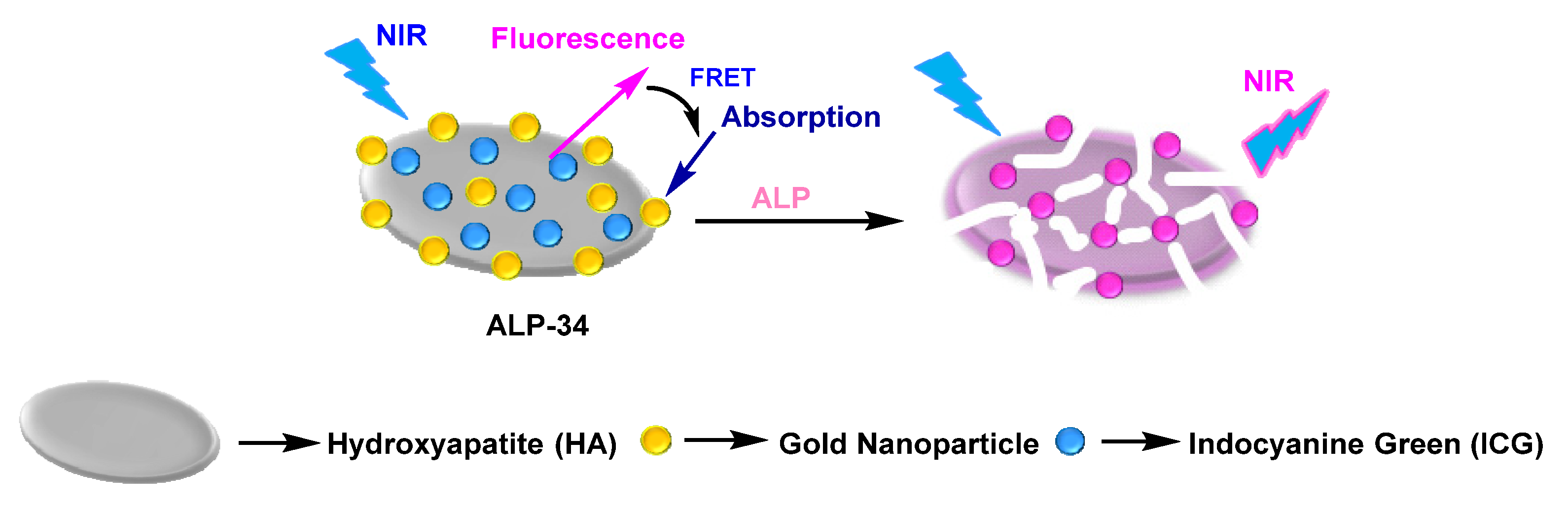
| ALP-34 | / | / | / | / | / | / | / | / | Turn-on fluorescence (NIR fluorescence) |
In vitro and live cells |
/ | Osteoblast | [100] |
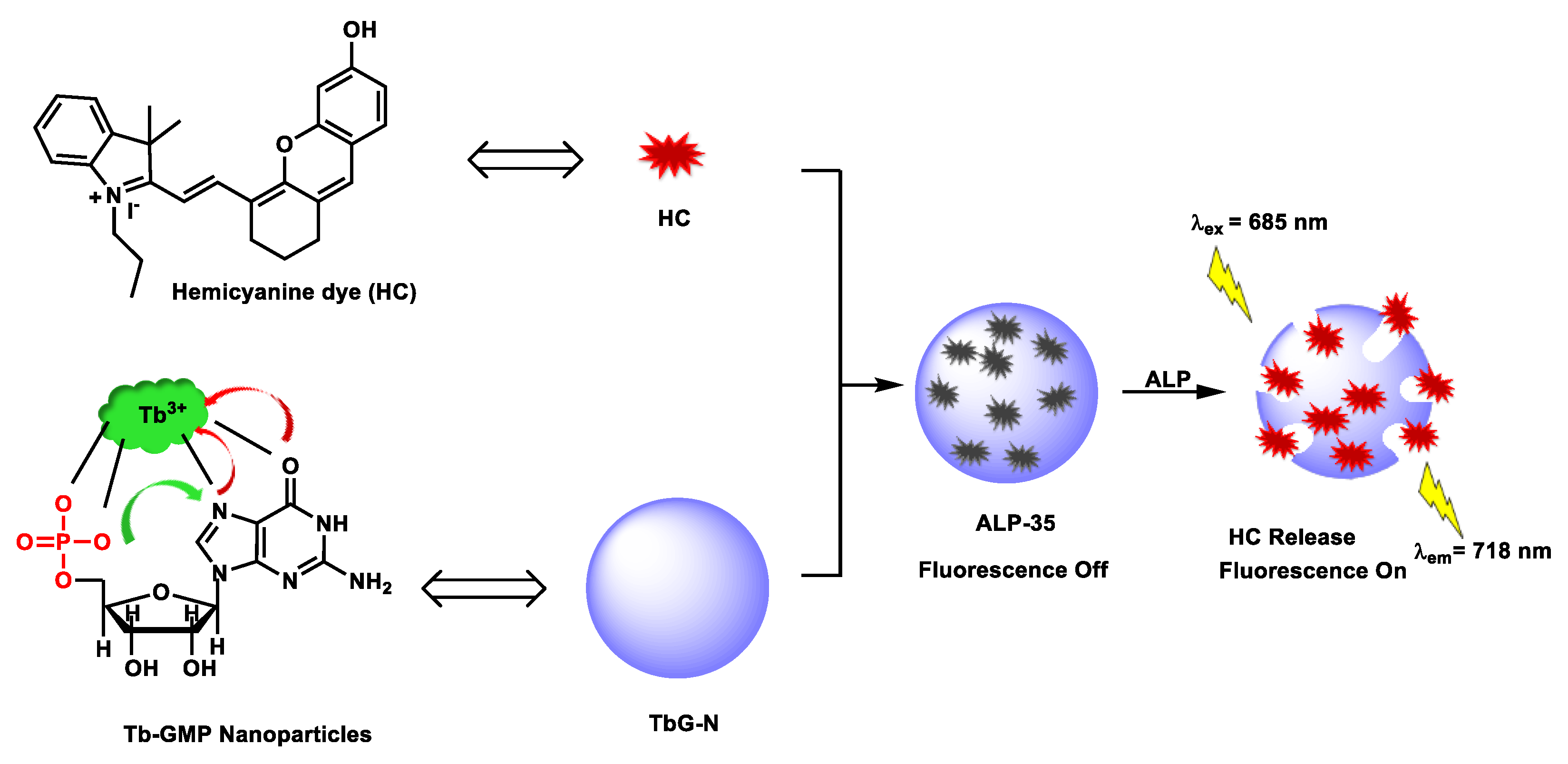
| ALP-35 | 0.01–2.5 U/mL | / | 0.0033 U/mL | / | / | 685/718 | / | 148.6 µM | Turn-on fluorescence (NIR fluorescence) |
In vivo | / | / | [101] |
4. Organo-Metallic-Based Fluorescent Probes
5. Nanomaterial-Based Fluorescent Probes
6. Miscellaneous
References
- Liang, J.; Kwok, R.T.K.; Shi, H.; Tang, B.Z.; Liu, B. Fluorescent Light-up Probe with Aggregation-Induced Emission Characteristics for Alkaline Phosphatase Sensing and Activity Study. ACS Appl. Mater. Interfaces 2013, 5, 8784–8789.
- McComb, R.B.; Bowers, G.N., Jr.; Posen, S. Alkaline Phosphatase, 1st ed.; Springer: New York, NY, USA; Plenum Press: New York, NY, USA, 1979.
- Yu, L.; Feng, L.; Xiong, L.; Li, S.; Xu, Q.; Pan, X.; Xiao, Y. Rational design of dual-emission lanthanide metal-organic framework for visual alkaline phosphatase activity assay. ACS Appl. Mater. Interfaces 2021, 13, 11646–11656.
- Ma, F.; Liu, M.; Zhang, C.-Y. Ligase amplification reaction-catalyzed assembly of a single quantum dot-based nanosensor for sensitive detection of alkaline phosphatase. Chem. Commun. 2019, 55, 8963–8966.
- Haarhaus, M.; Brandenburg, V.; Kalantar-Zadeh, K.; Stenvinkel, P.; Magnusson, P. Alkaline phosphatase: A novel treatment target for cardiovascular disease in CKD. Nat. Rev. Nephrol. 2017, 13, 429–442.
- Zhang, W.; Gao, Y.; Li, Y.; Zhang, Q.; Hu, Z.; Zhang, Y.; Hussain, E.; Yang, X.; Yu, D.; Yu, C. Polyphosphoric acid-induced perylene probe self-assembly and label-free fluorescence turn-on detection of alkaline phosphatase. Anal. Bioanal. Chem. 2017, 409, 1031–1036.
- Yang, D.; Guo, Z.; Tang, Y.; Miao, P. Poly(thymine)-templated selective formation of copper nanoparticles for alkaline phosphatase analysis aided by alkyne-azide cycloaddition “Click” reaction. ACS Appl. Nano Mater. 2018, 1, 168–174.
- Liu, X.-G.; Xing, X.-J.; Li, B.; Guo, Y.-M.; Zhang, Y.-Z.; Yang, Y.; Zhang, L.-F. Fluorescent assay for alkaline phosphatase activity based on graphene oxide integrating with λ exonuclease. Biosens. Bioelectron. 2016, 81, 460–464.
- Hu, X.-L.; Wu, X.-M.; Fang, X.; Li, Z.-J.; Wang, G.-L. Switchable fluorescence of gold nanoclusters for probing the activity of alkaline phosphatase and its application immunoassay. Biosens. Bioelectron. 2016, 77, 666–672.
- Liu, S.; Pang, S.; Na, W.; Su, X. Near-infrared fluorescence probe for the determination of alkaline phosphatase. Biosens. Bioelectron. 2014, 55, 249–254.
- Halawa, M.I.; Gao, W.; Saqib, M.; Kitte, S.A.; Wu, F.; Xu, G. Sensitive detection of alkaline phosphatase by switching on gold nanoclusters fluorescence quenched by pyridoxal phosphate. Biosens. Bioelectron. 2017, 95, 8–14.
- Rao, G.M.M.; Morghom, L.O. Correlation between serum alkaline phosphatase activity and blood glucose levels. Enzyme 1986, 35, 57–59.
- Sharma, U.; Pal, D.; Prasad, R. Alkaline Phosphatase: An Overview. Ind. J. Clin. Biochem. 2014, 29, 269–278.
- Wang, X.; Zhang, Z.; Ma, X.; Wen, J.; Geng, Z.; Wang, Z. Real-time fluorescence assays of alkaline phosphatase and ATP sulfurylase activities based on a novel PPi fluorescent probe. Talanta 2015, 137, 156–160.
- Deng, J.; Yu, P.; Wang, Y.; Mao, L. Real-time ratiometric fluorescent assay for alkaline phosphatase activity with stimulus responsive infinite coordination polymer nanoparticles. Anal. Chem. 2015, 87, 3080–3086.
- Zheng, F.; Guo, S.; Zeng, F.; Li, J.; Wu, S. Ratiometric fluorescent probe for alkaline phosphatase based on betaine-modified polyethylenimine via excimer/monomer conversion. Anal. Chem. 2014, 86, 9873–9879.
- Zhang, L.; Zhao, J.; Duan, M.; Zhang, H.; Jiang, J.; Yu, R. Inhibition of dsDNA-templated copper nanoparticles by pyrophosphate as a label-free fluorescent strategy for alkaline phosphatase assay. Anal. Chem. 2013, 85, 3797–3801.
- Wang, H.B.; Li, Y.; Chen, Y.; Zhang, Z.P.; Gan, T.; Liu, Y.M. Determination of the activity of alkaline phosphatase by using nanoclusters composed of flowerlike cobalt oxyhydroxide and copper nanoclusters as fluorescent probes. Microchim. Acta 2018, 185, 102.
- Li, G.; Huili Fu, H.; Chen, X.; Gong, P.; Chen, G.; Xia, L.; Wang, H.; You, J.; Wu, Y. Facile and sensitive fluorescence sensing of alkaline phosphatase activity with photoluminescent carbon dots based on inner filter effect. Anal. Chem. 2016, 88, 2720–2726.
- Cox, W.G.; Singer, V.L. A High-resolution, fluorescence-based method for localization, of endogenous alkaline phosphatase activity. J. Histochem. Cytochem. 1999, 47, 1443–1455.
- Butterworth, P.J. Biochemistry of alkaline phosphatases. Cell Biochem. Funct. Biochem. Data 1983, 1, 66–69.
- Štefková, K.; Procházková, J.; Pacherník, J. Alkaline phosphatase in stem cells. Stem Cells Int. 2015, 2015, 628368.
- Millan, J.L. Alkaline Phosphatases Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341.
- Chen, S.L.; Liao, R.Z. Phosphate monoester hydrolysis by trinuclear alkaline phosphatase; DFT study of transition states and reaction mechanism. ChemPhysChem 2014, 15, 2321–2330.
- Holtz, K.M.; Kantrowitz, E.R. The mechanism of the alkaline phosphatase reaction: Insights from NMR, crystallography and site-specific mutagenesis. FEBS Lett. 1999, 462, 7–11.
- Holtz, K.M.; Stec, B.; Kantrowitz, E.R. A model of the transition state in the alkaline phosphatase reaction. J. Biol. Chem. 1999, 274, 8351–8354.
- Chaudhuri, G.; Selvaraj, U.; Babu, V.; Thilagaraj, R.W.. Recent Trends in Phosphatase-Mediated Bioremediation; In Phosphoric Acid Industry Problems and Solutions; IntechOpen: London, UK, 2017; pp. 27–46.
- W Meyer-Sabellek; P Sinha; E Köttgen; Alkaline phosphatase. Laboratory and clinical implications.. Journal of chromatography 1988, 429, 419-44.
- Krupaa, R.J.; Hariharan, R.; Babu, N.A.; Masthan, K.M.K. Alkaline phosphatase and its clinical importance-A review. Eur. J. Mol. Clin. Med. 2020, 7, 1409–1413.
- Thanih Balbaied, T.; Moore, E. Overview of optical and electrochemical alkaline phosphatase (ALP) biosensors: Recent approaches in cells culture techniques. Biosensors 2019, 9, 102.
- Nsabimana, A.; Lan, Y.; Du, F.; Wang, C.; Zhang, W.; Xu, G. Alkaline phosphatase-based electrochemical sensors for health applications. Anal. Methods 2019, 11, 1996–2006.
- Goggins, S.; Naz, C.; Marsh, B.J.; Frost, C.G. Ratiometric electrochemical detection of alkaline phosphatase. Chem. Commun. 2015, 51, 561–564.
- Miao, P.; Ning, L.; Li, X.; Shu, Y.; Li, G. An electrochemical alkaline phosphatase biosensor fabricated with two DNA probes coupled with lambda exonuclease. Biosens. Bioelectron. 2011, 27, 178–182.
- Kim, H.-U.; Kim, H.Y.; Kulkarni, A.; Ahn, C.; Jin, Y.; Kim, Y.; Lee, K.-N.; Lee, M.-H.; Kim, T. A sensitive electrochemical sensor for in vitro detection of parathyroid hormone based on a MoS2-graphene composite. Sci. Rep. 2016, 6, 34587.
- Sun, D.; Xu, W.; Liang, C.; Shi, W.; Xu, S. Smart surface-enhanced resonance Raman scattering nanoprobe for monitoring cellular alkaline phosphatase activity during osteogenic differentiation. ACS Sens. 2020, 5, 1758–1767.
- Dai, X.; Lu, L.; Zhang, X.; Song, Z.-L.; Song, W.; Chao, Q.; Li, Q.; Wang, W.; Chen, J.; Fan, G.C.; et al. MnO2 shell-isolated SERS nanoprobe for the quantitative detection of ALP activity in trace serum: Relying on the enzyme-triggered etching of MnO2 shell to regulate the signal. Sens. Actuators B Chem. 2021, 334, 129605.
- Yi-Wei, T.; Charles, W.S. Advanced Techniques in Diagnostic Microbiology, 2nd ed.; Springer: New York, NY, USA, 2013.
- Roelofs, H.; Manes, T.; Janszen, T.; Millan, J.L.; Oosterhuis, J.W.; Looijenga, L.H.J. Heterogeneity in alkaline phosphatase isozyme expression in human testicular germ cell tumours: An enzyme-/immunohistochemical and molecular analysis. J. Pathol. 1999, 189, 236–244.
- Mano, H.; Furuhashi, Y.; Morikawa, Y.; Hattori, S.E.; Goto, S.; Tomoda, Y. Radioimmunoassay of placental alkaline-phosphatase in ovarian-cancer sera and tissues. Obstet. Gynecol. 1986, 68, 759–764.
- Degroote, G.; Dewaele, P.; Vandevoorde, A.; Debroe, M.; Fiers, W. Use of monoclonal-antibodies to detect human placental alkaline-phosphatase. Clin. Chem. 1983, 29, 115–119.
- Sekar, S.; Giermanska, J.; Chapel, J.P. Reusable and recyclable quartz crystal microbalance sensors. Sens. Actuators B Chem. 2015, 212, 196–199.
- Kacar, T.; Zin, M.T.; So, C.; Wilson, B.; Ma, H.; Gul-Karaguler, N.; Tamerler, C. Directed self-immobilization of alkaline phosphatase on micro-patterned substrates via genetically fused metal-binding peptide. Biotechnol. Bioeng. 2009, 103, 696–705.
- Jang, H.J.; Ahn, J.; Kim, M.G.; Shin, Y.B.; Jeun, M.; Cho, W.J.; Lee, K.H. Electrical signaling of enzyme-linked immunosorbent assays with an ion-sensitive field-effect transistor. Biosens. Bioelectron. 2015, 64, 318–323.
- Li, C.M.; Zhen, S.J.; Wang, J.; Li, Y.F.; Huang, C.Z. A gold nanoparticles-based colorimetric assay for alkaline phosphatase detection with tunable dynamic range. Biosens. Bioelectron. 2013, 43, 366–371.
- Hu, Q.; He, M.; Mei, Y.; Feng, W.; Jing, S.; Kong, J.; Zhang, X. Sensitive and selective colorimetric assay of alkaline phosphatase activity with Cu(II)-phenanthroline complex. Talanta 2017, 163, 146–152.
- Hu, X.; Sun, C.; Shi, Y.; Long, Y.; Zheng, H. Colorimetric sensing of alkaline phosphatase and a-fetoprotein based on the photoinduced oxidase activity of fluorescein. New J. Chem. 2019, 43, 4525–4530.
- Upadhyay, Y.; Kumar, R.; Sahoo, S.K. Developing a cost-effective bioassay to detect alkaline phosphatase activity and generating white light emission from a single nano-assembly by conjugating Vitamin B6 cofactors with lysozyme-stabilized fluorescent gold nanoclusters. ACS Sustain. Chem. Eng. 2020, 8, 4107–4113.
- Westmeyer, G.G.; Emer, Y.; Lintelmann, J.; Jasanoff, A. MRI-based detection of alkaline phosphatase gene reporter activity using a porphyrin solubility switch. Chem. Biol. 2014, 21, 422–429.
- Zhang, Q.; Li, S.; Fu, C.; Xiao, Y.; Zhang, P.; Ding, C. Near-infrared mito-specific fluorescent probe for ratiometric detection and imaging of alkaline phosphatase activity with high sensitivity. J. Mater. Chem. B 2019, 7, 443–445.
- Pandith, A.; Seo, Y.J. Label-free sensing platform for miRNA-146a based on chromo-fluorogenic pyrophosphate recognition. J. Inorg. Biochem. 2020, 203, 110867.
- Pandith, A.; Koo, J.; Seo, Y.J. Daphnetin: A novel blue-green photonic switch for disodium phosphates that allows monitoring of polymerase chain reactions. Spectrochim. Acta Part A 2018, 204, 620–628.
- Pandith, A.; Siddappa, R.G.; Seo, Y.J. Recent developments in novel blue/green/red/NIR small fluorescent probes for in cellulo tracking of RNA/DNA G-quadruplexes. J. Photochem. Photobiol. C Rev. 2019, 40, 81–116.
- Zhang, H.; Ju, Q.; Pang, S.; Wei, N.; Zhang, Y. Recent progress of fluorescent probes for the detection of alkaline phosphatase (ALP): A review. Dyes Pigments 2021, 194, 109569.
- Wang, K.; Wang, W.; Zhang, X.Y.; Jiang, A.Q.; Yang, Y.S.; Zhu, H.L. Fluorescent probes for the detection of alkaline phosphatase in biological systems: Recent advances and future prospects. Trends Anal. Chem. 2021, 136, 116189.
- Gwynne, L.; Sedgwick, A.C.; Gardiner, J.E.; Williams, G.T.; Kim, G.; Lowe, J.P.; Maillard, J.Y.; Jenkins, A.T.A.; Bull, S.D.; Sessler, J.L.; et al. Long wavelength TCF-based fluorescent probe for the detection of alkaline phosphatase in live cells. Front. Chem. 2019, 7, 255.
- Jidong, Z.; Hongze, L.; Li, M. Research Progress in the Fluorescent Probes for Alkaline Phosphatase. Chin. J. Org. Chem. 2019, 39, 3132–3144.
- Li, M.; Gurram, B.; Lei, S.; Blum, N.T.; Huang, P.; Lin, J. Recent advances in fluorescence imaging of alkaline phosphatase. Chin. Chem. Lett. 2021, 32, 1316–1330.
- Liu, H.W.; Chen, L.; Xu, C.; Li, Z.; Zhang, H.; Zhang, X.B.; Tan, W. Recent progresses in small-molecule enzymatic fluorescent probes for cancer imaging. Chem. Soc. Rev. 2018, 47, 7140–7180.
- Niu, X.; Ye, K.; Wang, L.; Lin, Y.; Du, D. A review on emerging principles and strategies for colorimetric and fluorescent detection of alkaline phosphatase activity. Anal. Chim. Acta 2019, 1086, 29–45.
- Han, Y.; Chen, J.; Li, Z.; Chen, H.; Qiu, H. Recent progress and prospects of alkaline phosphatase biosensor based on fluorescence strategy. Biosens. Bioelectron. 2020, 148, 111811.
- Hu, L.; Zhang, Q.; Gan, X.; Yin, W.; Fu, W. Switchable fluorescence of MoS2 quantum dots: A multifunctional probe for sensing of chromium (VI), ascorbic acid, and alkaline phosphatase activity. Anal. Bioanal. Chem. 2018, 410, 7551–7557.
- Syahir, A.; Usui, K.; Tomizaki, K.Y.; Kajikawa, K.; Mihara, H. Label and label-free detection techniques for protein microarrays. Microarrays 2015, 4, 228–244.
- Boaro, A.; Ageitos, L.; Torres, M.; Bartoloni, F.H.; de la Fuente-Nunez, C. Light-emitting probes for labeling peptides. Cell Rep. Phys. Sci. 2020, 1, 100257.
- Upadhyay, Y.; Kumar, R.; Sk, A.K.; Sahoo, S.K. Vitamin B6 cofactors conjugated ovalbumin-stabilized gold nanoclusters: Application in alkaline phosphatase activity detection and generating white-light emission. Microchem. J. 2020, 156, 104859.
- Upadhyay, Y.; Bothra, S.; Kumar, R.; Sk, A.K.; Sahoo, S.K. Mimicking biological process to detect alkaline phosphatase activity using the vitamin B6 cofactor conjugated bovine serum albumin capped CdS quantum dots. Colloids Surf. B Biointerfaces 2020, 185, 110624.
- Wang, J.H.; Wang, K.; Bartling, B.; Liu, C.C. The detection of alkaline phosphatase using an electrochemical biosensor in a single-step approach. Sensors 2009, 9, 8709–8721.
- Li, X.; Wang, X.; Guo, W.; Wang, Y.; Hua, Q.; Tang, F.; Luan, F.; Tian, C.; Zhuang, X.; Zhao, L. Selective Detection of Alkaline Phosphatase Activity in Environmental Water Samples by Copper Nanoclusters Doped Lanthanide Coordination Polymer Nanocomposites as the Ratiometric Fluorescent Probe. Biosensors 2022, 12, 372.
- Cao, Y.; Yu, X.; Sun, C.; Cui, J. Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe. Int. J. Mol. Sci. 2022, 23, 2132.
- Sun, T.; Xia, N.; Liu, L. A graphene oxide-based fluorescent platform for probing of phosphatase activity. Nanomaterials 2016, 6, 20.
- David, C.I.; Prabakaran, G.; Nandhakumar, R. Recent approaches of 2HN derived fluorophores on recognition of Al3+ ions: A review for future outlook. Microchem. J. 2021, 169, 106590.
- Cao, F.Y.; Long, Y.; Wang, S.B.; Li, B.; Fan, J.X.; Zeng, X.; Zhang, X.Z. Fluorescence light-up AIE probe for monitoring cellular alkaline phosphatase activity and detecting osteogenic differentiation. J. Mater. Chem. B 2016, 4, 4534–4541.
- Song, Z.; Kwok, R.T.; Zhao, E.; He, Z.; Hong, Y.; Lam, J.W.; Liu, B.; Tang, B.Z. A ratiometric fluorescent probe based on ESIPT and AIE processes for alkaline phosphatase activity assay and visualization in living cells. ACS Appl. Mater. Interfaces 2014, 6, 17245–17254.
- Zhang, H.; Xiao, P.; Wong, Y.T.; Shen, W.; Chhabra, M.; Peltier, R.; Jiang, Y.; He, Y.; He, J.; Tan, Y.; et al. Construction of an alkaline phosphatase-specific two-photon probe and its imaging application in living cells and tissues. Biomaterials 2017, 140, 220–229.
- Li, S.J.; Li, C.Y.; Li, Y.F.; Fei, J.; Wu, P.; Yang, B.; Ou-Yang, J.; Nie, S.X. Facile and sensitive near-infrared fluorescence probe for the detection of endogenous alkaline phosphatase activity in vivo. Anal. Chem. 2017, 89, 6854–6860.
- Dong, L.; Miao, Q.; Hai, Z.; Yuan, Y.; Liang, G. Enzymatic hydrogelation-induced fluorescence turn-off for sensing alkaline phosphatase in vitro and in living cells. Anal. Chem. 2015, 87, 6475–6478.
- Tan, Y.; Zhang, L.; Man, K.H.; Peltier, R.; Chen, G.; Zhang, H.; Zhou, L.; Wang, F.; Ho, D.; Yao, S.Q.; et al. Reaction-based off–on near-infrared fluorescent probe for imaging alkaline phosphatase activity in living cells and mice. ACS Appl. Mater. Interfaces 2017, 9, 6796–6803.
- Jie, X.; Wu, M.; Yang, H.; Wei, W. Red–Near-Infrared Fluorescent Probe for Time-Resolved in Vivo Alkaline Phosphatase Detection with the Assistance of a Photoresponsive Nanocontainer. Anal. Chem. 2019, 91, 13174–13182.
- Zhang, P.; Fu, C.; Zhang, Q.; Li, S.; Ding, C. Ratiometric fluorescent strategy for localizing alkaline phosphatase activity in mitochondria based on the ESIPT process. Anal. Chem. 2019, 91, 12377–12383.
- Jia, Y.; Li, P.; Han, K. AMP/GMP analogs as affinity ESIPT probes for highly selective sensing of alkaline phosphatase activity in living systems. Chem. Asian J. 2015, 10, 2444–2451.
- Kim, T.I.; Kim, H.; Choi, Y.; Kim, Y. A fluorescent turn-on probe for the detection of alkaline phosphatase activity in living cells. Chem. Commun. 2011, 47, 9825–9827.
- Zhang, H.; Xu, C.; Liu, J.; Li, X.; Guo, L.; Li, X. An enzyme-activatable probe with a self-immolative linker for rapid and sensitive alkaline phosphatase detection and cell imaging through a cascade reaction. Chem. Commun. 2015, 51, 7031–7034.
- He, Y.; Yu, J.; Hu, X.; Huang, S.; Cai, L.; Yang, L.; Zhang, H.; Jiang, Y.; Jia, Y.; Sun, H. An activity-based fluorescent probe and its application for differentiating alkaline phosphatase activity in different cell lines. Chem. Commun. 2020, 56, 13323–13326.
- Hou, X.; Yu, Q.; Zeng, F.; Ye, J.; Wu, S. A ratiometric fluorescent probe for in vivo tracking of alkaline phosphatase level variation resulting from drug-induced organ damage. J. Mater. Chem. B 2015, 3, 1042–1048.
- Xiaohong Zhou; Yuren Jiang; Xiongjie Zhao; Yao Zhu; A New Two-Photon Ratiometric Fluorescent Probe for Detecting Alkaline Phosphatase in Living Cells. Molecules 2016, 21, 1619, 10.3390/molecules21121619.
- Podder, A.; Senapati, S.; Maiti, P.; Kamalraj, D.; Jaffer, S.S.; Khatun, S.; Bhuniya, S. A ‘turn-on’ fluorescent probe for lysosomal phosphatase: A comparative study for labeling of cancer cells. J. Mater. Chem. B 2018, 6, 4514–4521.
- Lu, Z.; Wu, J.; Liu, W.; Zhang, G.; Wang, P. A ratiometric fluorescent probe for quantification of alkaline phosphatase in living cells. RSC Adv. 2016, 6, 32046–32051.
- Hu, Q.; Zeng, F.; Yu, C.; Wu, S. A fluorescent probe for alkaline phosphatase via excited state intramolecular proton transfer. Sens. Actuators B Chem. 2015, 220, 720–726.
- Liu, H.W.; Hu, X.X.; Zhu, L.; Li, K.; Rong, Q.; Yuan, L.; Zhang, X.B.; Tan, W. In vivo imaging of alkaline phosphatase in tumor-bearing mouse model by a promising near-infrared fluorescent probe. Talanta 2017, 175, 421–426.
- Wang, W.X.; Jiang, W.L.; Guo, H.; Li, Y.; Li, C.Y. Real-time imaging of alkaline phosphatase activity of diabetes in mice via a near-infrared fluorescent probe. Chem. Commun. 2021, 57, 480–483.
- Pang, X.; Li, Y.; Lu, Q.; Ni, Z.; Zhou, Z.; Xie, R.; Wu, C.; Li, H.; Zhang, Y. A turn-on near-infrared fluorescent probe for visualization of endogenous alkaline phosphatase activity in living cells and zebrafish. Analyst 2021, 146, 521–528.
- Khatun, S.; Biswas, S.; Mahanta, A.K.; Joseph, M.M.; Vidyalekshmi, M.S.; Podder, A.; Maiti, P.; Maiti, K.K.; Bhuniya, S. Biocompatible fluorescent probe for detecting mitochondrial alkaline phosphatase activity in live cells. J. Photochem. Photobiol. B Biol. 2020, 212, 112043.
- Park, C.S.; Ha, T.H.; Kim, M.; Raja, N.; Yun, H.S.; Sung, M.J.; Kwon, O.S.; Yoon, H.; Lee, C.S. Fast and sensitive near-infrared fluorescent probes for ALP detection and 3d printed calcium phosphate scaffold imaging in vivo. Biosens. Bioelectron. 2018, 105, 151–158.
- Li, Y.; Xie, R.; Pang, X.; Zhou, Z.; Xu, H.; Gu, B.; Wu, C.; Li, H.; Zhang, Y. Aggregation-induced emission fluorescent probe for monitoring endogenous alkaline phosphatase in living cells. Talanta 2019, 205, 120143.
- Li, J.; Huo, F.; Wen, Z.; Yin, C. A fluorescent turn-on probe based on isophorone for the rapid detection of alkaline phosphatase and its application in bioimaging. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 221, 117156.
- Yangyang, Y.; Chen, Z.; Rizhao, P.; Shiwei, Z.; Shengtao, Y.; Yao, T.; Weilong, Z.; Liyue, W.; Weiping, Z.; Yufang, X.; et al. A ratiometric fluorescent probe for alkaline phosphatase with high sensitivity. Chin. Chem. Lett. 2020, 31, 125–128.
- Gao, C.; Zang, S.; Nie, L.; Tian, Y.; Zhang, R.; Jing, J.; Zhang, X. A sensitive ratiometric fluorescent probe for quantitative detection and imaging of alkaline phosphatase in living cells. Anal. Chim. Acta 2019, 1066, 131–135.
- Xu, L.; He, X.; Huang, Y.; Ma, P.; Jiang, Y.; Liu, X.; Tao, S.; Sun, Y.; Song, D.; Wang, X. A novel near-infrared fluorescent probe for detecting intracellular alkaline phosphatase and imaging of living cells. J. Mater. Chem. B 2019, 7, 1284–1291.
- Lin, M.; Huang, J.; Zeng, F.; Wu, S. A fluorescent probe with aggregation-induced emission for detecting alkaline phosphatase and cell imaging. Chem. Asian J. 2019, 14, 802–808.
- Pandith, A.; Bhattarai, K.R.; Siddappa, R.K.G.; Chae, H.J.; Seo, Y.J. Novel fluorescent C2-symmetric sequential on-off-on switch for Cu2+ and pyrophosphate and its application in monitoring of endogenous alkaline phosphatase activity. Sens. Actuators B Chem. 2019, 282, 730–742.
- Lim, E.K.; Keem, J.O.; Yun, H.S.; Jung, J.; Chung, B.H. Smart nanoprobes for the detection of alkaline phosphatase activity during osteoblast differentiation. Chem. Commun. 2015, 51, 3270–3272.
- Juan Ou-Yang; Chun-Yan Li; Yong-Fei Li; Bin Yang; Song-Jiao Li; An infinite coordination polymer nanoparticles-based near-infrared fluorescent probe with high photostability for endogenous alkaline phosphatase in vivo. Sensors and Actuators B: Chemical 2018, 255, 3355-3363, 10.1016/j.snb.2017.09.162.

