 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cenap Güngör | -- | 3506 | 2022-08-26 09:47:07 | | | |
| 2 | Lindsay Dong | -13 word(s) | 3493 | 2022-08-29 07:03:21 | | |
Video Upload Options
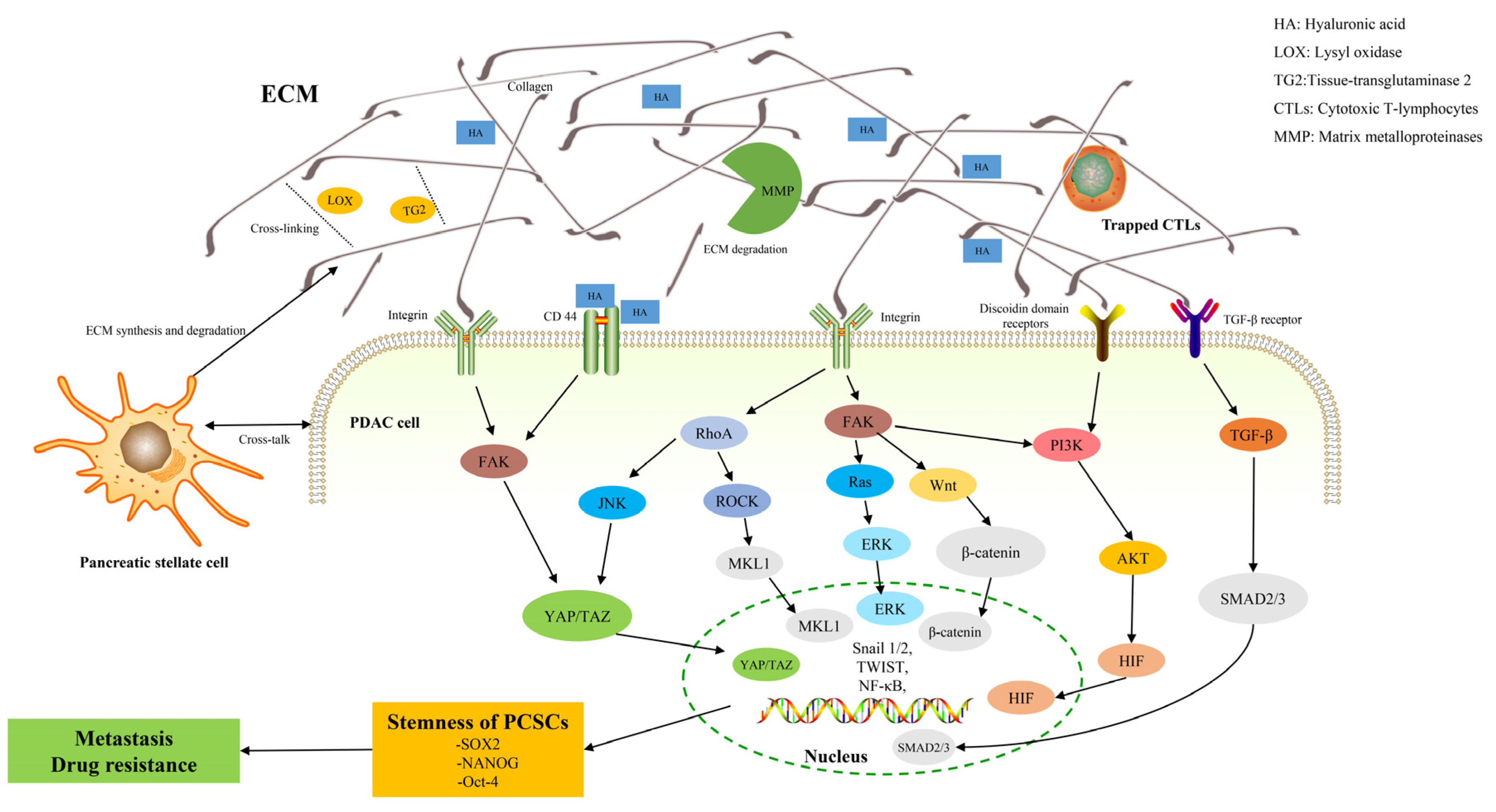
Pancreatic ductal adenocarcinoma (PDAC) is rich in dense fibrotic stroma that are composed of extracellular matrix (ECM) proteins. A disruption of the balance between ECM synthesis and secretion and the altered expression of matrix remodeling enzymes lead to abnormal ECM dynamics in PDAC. This pathological ECM promotes cancer growth, survival, invasion, and alters the behavior of fibroblasts and immune cells leading to metastasis formation and chemotherapy resistance, which contribute to the high lethality of PDAC. Additionally, recent evidence highlights that ECM, as a major structural component of the tumor microenvironment, is a highly dynamic structure in which ECM proteins establish a physical and biochemical niche for cancer stem cells (CSCs). CSCs are characterized by self-renewal, tumor initiation, and resistance to chemotherapeutics.
1. Introduction
2. ECM in PDAC
3. ECM and Pancreatic Cancer Stem Cells (PCSCs)
4. ECM, PCSCs, and Metastasis
5. ECM, PCSCs, and Chemoresistance
6. Pharmacological Targeting of ECM
Collagen is one of the most basic components in the ECM, and it is also one of the ideal therapeutic directions. TGF-β plays a key role in collagen synthesis, so TGF-β signaling is the most promising target for inhibiting collagen synthesis. In animal models of PDAC, an anticoccidial named halofuginone has been shown to reduce collagen synthesis by inhibiting TGF-β signaling [112]. TGF-β is usually overexpressed in PDAC and blocking TGF-β-mediated signaling may enhance antitumor effects [110][111]. A clinical trial aiming to target TGF-β in PDAC is investigating the antitumor activity of SAR439459, a pan-TGF-β neutralizing antibody [113]. Vitamin D may also block collagen secretion by disrupting the TGF-β signaling pathway, helping to prevent tumor metastasis and enhance drug responses [114][115]. The inhibition of collagen cross-linking is also a therapeutic strategy to target ECM stiffness in cancer. In 2017, a randomized Phase II study of Simtuzumab, a humanized IgG4 monoclonal antibody that inhibits extracellular LOXL2, was performed in combination with gemcitabine in patients that were suffering from metastatic PDAC [84]. However, the results showed that a combination of gemcitabine and Simtuzumab did not improve clinical outcomes. Therefore, the complex tumor-stromal interaction of LOXL2 in PDAC tumorigenesis requires further investigation and re-evaluation of the efficacy of using anti-LOXL2 antibodies or small molecule LOXL2 inhibitors in the treatment landscape of metastatic PDAC [116].
There are other therapeutic ideas aiming to target the MMP to alter ECM stiffness. For example, MMP-9 promotes aggressive and metastatic phenotypes in tumor cells, and its overexpression increases the aggressiveness of cancer cell lines in vitro [117]. Targeting integrin-mediated signaling is another strategy for ECM therapy. In theory, it is possible to disrupt signals from the extracellular or intracellular environment by using integrin inhibitors to disrupt ECM mechanical sensing [118]. Targeted glutamine metabolism and hexosamine biosynthesis pathways has been shown to have profound effects on the ECM and CSC self-renewal of PDAC among metabolic inhibitors [119].
Clinical trials involving different inhibitors are being conducted in different clinical settings, primarily for ECM-induced stemness of PCSCs. For example, the inhibitors of Rho-associated kinase (ROCK) alter the contractility of PDAC cytoskeleton and CAF, may benefit drug delivery and inhibit metastasis. ECM disorder occurrence and PDAC migration and invasion are prevented when ROCK is suppressed. Other mouse models showed that a pre-chemotherapy administration of ROCK inhibitors increased the chemotherapy response of the primary tumor and helped to prevent the development of liver metastases [120][121][122]. T13148 was the first ROCK inhibitor that was investigated for the treatment of solid tumors, but further development of this compound is advised due to drug side effects [123].
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800.
- Sally, A.; McGowan, R.; Finn, K.; Moran, B.M. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2417.
- Güngör, C.; Hofmann, B.T.; Wolters-Eisfeld, G.; Bockhorn, M. Pancreatic cancer. Br. J. Pharmacol. 2014, 171, 849–858.
- Balaban, E.P.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Mukherjee, S.; Crane, C.H.; Javle, M.M.; Eads, J.R.; Allen, P.; Ko, A.H.; et al. Locally Advanced, Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 2654–2668.
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327.
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346.
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471.
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812.
- Hellewell, A.L.; Adams, J.C. Insider trading: Extracellular matrix proteins and their non-canonical intracellular roles. Bioessays 2016, 38, 77–88.
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 57, 10.1.1–10.1.16.
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224.
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134.
- Tian, C.; Ohlund, D.; Rickelt, S.; Lidstrom, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474.
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120.
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801.
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153.
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67.
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087.
- Jamin, Y.; Boult, J.K.R.; Li, J.; Popov, S.; Garteiser, P.; Ulloa, J.L.; Cummings, C.; Box, G.; Eccles, S.A.; Jones, C.; et al. Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography. Cancer Res. 2015, 75, 1216–1224.
- Hu, G.; Xu, F.; Zhong, K.; Wang, S.; Huang, L.; Chen, W. Activated Tumor-infiltrating Fibroblasts Predict Worse Prognosis in Breast Cancer Patients. J. Cancer 2018, 9, 3736–3742.
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523.
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65.
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568.
- Apte, M.V.; Xu, Z.; Pothula, S.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Pancreatic cancer: The microenvironment needs attention too! Pancreatology 2015, 15, S32–S38.
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: A conspiracy theory. World J. Gastroenterol. 2014, 20, 11216–11229.
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556.
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86.
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347.
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904.
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123.
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058.
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901.
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706.
- Craven, K.E.; Gore, J.; Korc, M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 381, 201–210.
- Fukumura, D.; Jain, R.K. Imaging angiogenesis and the microenvironment. APMIS 2008, 116, 695–715.
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 2000, 1, 510–514.
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886.
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700.
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921.
- Tian, C.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618.
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24.
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867.
- Yang, C.; Liu, Z.; Zeng, X.; Wu, Q.; Liao, X.; Wang, X.; Han, C.; Yu, T.; Zhu, G.; Qin, W.; et al. Evaluation of the diagnostic ability of laminin gene family for pancreatic ductal adenocarcinoma. Aging 2019, 11, 3679–3703.
- Katayama, M.; Sekiguchi, K. Laminin-5 in epithelial tumour invasion. J. Mol. Histol. 2004, 35, 277–286.
- Wang, H.; Fu, W.; Im, J.H.; Zhou, Z.; Santoro, S.A.; Iyer, V.; DiPersio, C.M.; Yu, Q.C.; Quaranta, V.; Al-Mehdi, A.; et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J. Cell Biol. 2004, 164, 935–941.
- Ortiz-Urda, S.; Garcia, J.; Green, C.L.; Chen, L.; Lin, Q.; Veitch, D.P.; Sakai, L.Y.; Lee, H.; Marinkovich, M.P.; Khavari, P.A. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science 2005, 307, 1773–1776.
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 2011, 4, 21.
- Zeltz, C.; Orgel, J.; Gullberg, D. Molecular composition and function of integrin-based collagen glues-introducing COLINBRIs. Biochim. Biophys. Acta 2014, 1840, 2533–2548.
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44.
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778.
- Chen, R.; Dawson, D.W.; Pan, S.; Ottenhof, N.A.; de Wilde, R.F.; Wolfgang, C.L.; May, D.H.; Crispin, D.A.; Lai, L.A.; Lay, A.R.; et al. Proteins associated with pancreatic cancer survival in patients with resectable pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 43–55.
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319.
- Theocharis, A.D.; Tsara, M.E.; Papageorgacopoulou, N.; Karavias, D.D.; Theocharis, D.A. Pancreatic carcinoma is characterized by elevated content of hyaluronan and chondroitin sulfate with altered disaccharide composition. Biochim. Biophys. Acta 2000, 1502, 201–206.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429.
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4.
- Mahlbacher, V.; Sewing, A.; Elsasser, H.P.; Kern, H.F. Hyaluronan is a secretory product of human pancreatic adenocarcinoma cells. Eur. J. Cell Biol. 1992, 58, 28–34.
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e21.
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344.
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39.
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311.
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254.
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732.
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin. Cancer Res. 2015, 21, 4482–4493.
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352.
- Haage, A.; Schneider, I.C. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014, 28, 3589–3599.
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037.
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344.
- Motegi, H.; Kamoshima, Y.; Terasaka, S.; Kobayashi, H.; Houkin, K. Type 1 collagen as a potential niche component for CD133-positive glioblastoma cells. Neuropathology 2014, 34, 378–385.
- Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067.
- Grassian, A.R.; Coloff, J.L.; Brugge, J.S. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 313–324.
- Vitale, D.; Kumar Katakam, S.; Greve, B.; Jang, B.; Oh, E.S.; Alaniz, L.; Gotte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882.
- Wu, C.; Zhang, H.F.; Gupta, N.; Alshareef, A.; Wang, Q.; Huang, Y.H.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Lai, R. A positive feedback loop involving the Wnt/beta-catenin/MYC/Sox2 axis defines a highly tumorigenic cell subpopulation in ALK-positive anaplastic large cell lymphoma. J. Hematol. Oncol. 2016, 9, 120.
- Munir, J.; Van Ngu, T.; Na Ayudthaya, P.D.; Ryu, S. Downregulation of glypican-4 facilitates breast cancer progression by inducing cell migration and proliferation. Biochem. Biophys. Res. Commun. 2020, 526, 91–97.
- Zhao, D.; Liu, S.; Sun, L.; Zhao, Z.; Liu, S.; Kuang, X.; Shu, J.; Luo, B. Glypican-4 gene polymorphism (rs1048369) and susceptibility to Epstein-Barr virus-associated and -negative gastric carcinoma. Virus Res. 2016, 220, 52–56.
- Skandalis, S.S.; Karalis, T.T.; Chatzopoulos, A.; Karamanos, N.K. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell. Signal. 2019, 63, 109377.
- Porsch, H.; Bernert, B.; Mehic, M.; Theocharis, A.D.; Heldin, C.H.; Heldin, P. Efficient TGFbeta-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 2013, 32, 4355–4365.
- Preca, B.T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577.
- Steinbichler, T.B.; Savic, D.; Dudas, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156.
- Kim, Y.M.; Kim, E.C.; Kim, Y. The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol. Biol. Rep. 2011, 38, 145–149.
- Moon, H.J.; Finney, J.; Ronnebaum, T.; Mure, M. Human lysyl oxidase-like 2. Bioorg. Chem. 2014, 57, 231–241.
- Cano, A.; Santamaria, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 2012, 8, 1095–1108.
- Alonso-Nocelo, M.; Ruiz-Canas, L.; Sancho, P.; Gorgulu, K.; Alcala, S.; Pedrero, C.; Vallespinos, M.; Lopez-Gil, J.C.; Ochando, M.; Garcia-Garcia, E.; et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut 2022.
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874.
- Jiang, W.; Zhang, Y.; Kane, K.T.; Collins, M.A.; Simeone, D.M.; di Magliano, M.P.; Nguyen, K.T. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol. Cancer Res. 2015, 13, 9–15.
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Duraes, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via CCL18. Biomaterials 2017, 124, 211–224.
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910.
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160.
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268.
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761.
- Elia, I.; Doglioni, G.; Fendt, S.M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684.
- Attanasio, F.; Caldieri, G.; Giacchetti, G.; van Horssen, R.; Wieringa, B.; Buccione, R. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 2011, 90, 115–127.
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312.
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201.
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227.
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674.
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279.
- Zhang, B.; Wang, H.; Jiang, T.; Jin, K.; Luo, Z.; Shi, W.; Mei, H.; Wang, H.; Hu, Y.; Pang, Z.; et al. Cyclopamine treatment disrupts extracellular matrix and alleviates solid stress to improve nanomedicine delivery for pancreatic cancer. J. Drug Target. 2018, 26, 913–919.
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528.
- Du Souich, P.; Fradette, C. The effect and clinical consequences of hypoxia on cytochrome P450, membrane carrier proteins activity and expression. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1083–1100.
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394.
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316.
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291.
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK-PI3K/Akt pathway. Cell Biol. Int. 2014, 38, 85–91.
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515.
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37.
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976.
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget 2016, 7, 36719–36732.
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976.
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8.
- Zion, O.; Genin, O.; Kawada, N.; Yoshizato, K.; Roffe, S.; Nagler, A.; Iovanna, J.L.; Halevy, O.; Pines, M. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas 2009, 38, 427–435.
- Greco, R.; Qu, H.; Qu, H.; Theilhaber, J.; Shapiro, G.; Gregory, R.; Winter, C.; Malkova, N.; Sun, F.; Jaworski, J.; et al. Pan-TGFbeta inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. Oncoimmunology 2020, 9, 1811605.
- Shany, S.; Sigal-Batikoff, I.; Lamprecht, S. Vitamin D and Myofibroblasts in Fibrosis and Cancer: At Cross-purposes with TGF-beta/SMAD Signaling. Anticancer Res. 2016, 36, 6225–6234.
- Artaza, J.N.; Norris, K.C. Vitamin D reduces the expression of collagen and key profibrotic factors by inducing an antifibrotic phenotype in mesenchymal multipotent cells. J. Endocrinol. 2009, 200, 207–221.
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078.
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238.
- Rezza, A.; Sennett, R.; Rendl, M. Adult stem cell niches: Cellular and molecular components. Curr. Top. Dev. Biol. 2014, 107, 333–372.
- Sharma, N.S.; Gupta, V.K.; Garrido, V.T.; Hadad, R.; Durden, B.C.; Kesh, K.; Giri, B.; Ferrantella, A.; Dudeja, V.; Saluja, A.; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Investig. 2020, 130, 451–465.
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9, eaai8504.
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiello, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho Kinase Inhibition by AT13148 Blocks Pancreatic Ductal Adenocarcinoma Invasion and Tumor Growth. Cancer Res. 2018, 78, 3321–3336.
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2020, 11, 45–52.
- McLeod, R.; Kumar, R.; Papadatos-Pastos, D.; Mateo, J.; Brown, J.S.; Garces, A.H.I.; Ruddle, R.; Decordova, S.; Jueliger, S.; Ferraldeschi, R.; et al. First-in-Human Study of AT13148, a Dual ROCK-AKT Inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2020, 26, 4777–4784.

