Pancreatic ductal adenocarcinoma (PDAC) is rich in dense fibrotic stroma that are composed of extracellular matrix (ECM) proteins. A disruption of the balance between ECM synthesis and secretion and the altered expression of matrix remodeling enzymes lead to abnormal ECM dynamics in PDAC. This pathological ECM promotes cancer growth, survival, invasion, and alters the behavior of fibroblasts and immune cells leading to metastasis formation and chemotherapy resistance, which contribute to the high lethality of PDAC. Additionally, recent evidence highlights that ECM, as a major structural component of the tumor microenvironment, is a highly dynamic structure in which ECM proteins establish a physical and biochemical niche for cancer stem cells (CSCs). CSCs are characterized by self-renewal, tumor initiation, and resistance to chemotherapeutics.
- pancreatic ductal adenocarcinoma
- extracellular matrix
- cancer stem cells
- chemotherapy resistance
- metastasis
1. Introduction
2. ECM in PDAC
Abnormal remodeling of the ECM is associated with a variety of pathologic conditions, such as inflammation, fibrosis, and cancer [16][40]. In particular, a growing number of studies have highlighted the active role of dysregulated ECM dynamics in tumor progression and invasion [17][18][18,44]. The expression of many ECM remodeling enzymes is frequently deregulated in human cancers, most notably Matrix metalloproteinases (MMPs) [19][45]. MMPs are secreted by cancer-associated fibroblasts (CAFs) in PDAC [20][46]. Fibroblasts in the tumor microenvironment, collectively referred to as CAFs, are associated with tumor aggressiveness and reduced survival [21][22][47,48]. The CAF population, due to the different cellular origins, exhibits marked heterogeneity and functional diversity [23][49]. One of the most common cellular origins of CAFs is pancreatic stellate cells (PSCs) [24][50]. After pancreatic injury, or TGF-β stimulation, PSCs were activated, resulting in changes in cell morphology from stellate to spindle, increased nuclear volume, and decreased vitamin A droplets [25][26][27][51,52,53]. CAFs can also originate from tissue-resident fibroblasts, which can be activated under the control of growth factors (such as TGF-β) [28][54]. Additional studies have demonstrated that CAFs may also arise from trans-differentiation of non-fibroblast lineages or epithelial cells, as well as the recruitment and differentiation of bone marrow-derived mesenchymal stem cells [20][29][46,55]. Markers that were used to distinguish different CAF subsets include α-SMA, platelet-derived growth factor receptors α and β (PDGFRα/β), FAP, and Ca2+ binding protein (S100A4), but none of them are exclusively expressed by CAFs [29][30][55,56]. In terms of functional diversity, CAFs can secrete MMP, which promotes the degradation of the ECM and the release of various factors, and leads to the recruitment of some specific cells and various cytokines. On the other hand, CAFs are mainly responsible for the deposition of dense tumor stroma, which can serve as a structural scaffold for cell interactions and a physical barrier against immune infiltration. Furthermore, many growth factors and proinflammatory cytokines, including interleukin 6 (IL-6), TGF-β, and vascular endothelial growth factor (VEGF) can be produced by CAFs, which can promote tumor growth, angiogenesis, and assists in immune escape [20][31][46,57]. Therefore, CAFs, combining the above characteristics, are often programmed into three different subgroups: “antigen-presenting”, “myofibroblastic”, and “inflammatory” CAFs [32][58]. The dysregulation of ECM remodeling can cause mutant cells to escape apoptosis due to the pro- and anti-apoptotic effects of various ECM components or their functional fragments [33][59]. Abnormal ECM remodeling affects the behavior of not only cancer cells, but also those of stromal, endothelial, and immune cells of the local microenvironment as well [34][35][60,61]. Angiogenesis that is induced by abnormal ECM remodeling often results in an abnormal vasculature that is characterized by tortuous and immature leaky vessels in malignancies [36][62]. Vascular abnormalities, including the absence of a lymphatic network within the tumor, lead to increased interstitial fluid pressure, acidosis, and hypoxia, which underlie metastasis and drug resistance [37][63]. IL-6 that is secreted by stromal CAFs recruits tumor-associated macrophages and promotes their transition to an immunosuppressive phenotype (M2) [38][64]. Stromal CAFs recruit and induce Treg differentiation to suppress antitumor immunity and similarly enhance the tumor-promoting function of CD4+ helper T (Th) lymphocytes [39][65]. A prominent feature of PDAC is a strong pro-fibrotic response leading to a hyper-fibrotic stroma. Sparse vascularization and abundant deposition of extracellular components lay the groundwork for the stroma of PDAC [40][67]. Collagen, produced mainly by CAFs (especially derived from PSCs), is the most representative component of PDAC connective tissue [41][68]. Fibrinous collagen Type I and III account for more than 90% of the total and protein levels of these collagen proteins that increased nearly three-fold during the progression of PDAC [42][69]. However, compared with normal tissue, the ratio of these two kinds of collagen in tumor connective tissue did not change significantly, so one should pay attention to other differentially expressed collagen in the process of PDAC [43][70]. Proteoglycans and glycoproteins are composed of core proteins that undergo post-translational glycosylation, which largely shapes their conformation and cell signaling functions, and has important effects on tumor cells [44][77]. Most subunits of laminin, a small leucine-rich proteoglycan, are overexpressed in PDAC and are related to poor prognosis [45][78]. In particular, Lm332 and Lm511/521, which are currently common in various tumors, promote tumor progression by interacting with other proteins and cytokines [46][33]. The interaction of Lm332 with collagen Type VII and integrin α3β1 promotes tumor cell growth and metastasis, respectively [47][48][79,80]. Fibronectin has been shown to act as scaffolds and regulate cellular processes in the PDAC microenvironment [49][81]. Fibronectin stimulates tumor cell proliferation and invasion of the basement membrane and acts as a bridging molecule between ECM collagen and integrin [50][51][82,83]. Moreover, studies have shown that reduced galectin-1 (GAL1) in mouse models leads to reduced matrix activation and increased cytotoxic T-cell infiltration [52][84]. GAL1, as a class of glycoproteins, is expressed in multiple tumor types and is involved in proliferation, invasion, angiogenesis, metastasis, and is associated with patient survival [53][54][85,86]. Compared with normal pancreas, the total glycosaminoglycan content was significantly increased in PDAC (four-fold, p ≤ 0.001), mainly due to increased hyaluronic acid content [55][96]. A large amount of hyaluronic acid is already produced in the tumor microenvironment in the pre-cancerous intraepithelial neoplasia (PanIN) stage [56][97]. Hyaluronic acid is thought to be deposited primarily by CAFs and to some extent by PDAC cells [57][58][98,99]. Most importantly, hyaluronidase breaks down the hyaluronic acid matrix to allow interactions between growth factors and growth factor receptors, promoting glucose metabolism, tumor cell proliferation, and migration [59][100]. PSCs are key to maintaining the balance between ECM synthesis and degradation, and most studies showed an “activated” state in PDAC, leading to excessive deposition of ECM proteins [60][101]. It also leads to the increased expression of collagen, αSMA, immunomodulatory and other pro-tumorigenic genes [60][101]. TGF-β promotes ECM deposition and tumor progression in late stages of carcinogenesis [61][102]. As a potent activator of PSC, TGF-β mediates the interaction between the tumor microenvironment and tumor cells by binding to TGF-β cell surface receptors, promoting the deposition of ECM proteins including fibronectin and collagen [62][103]. The PSCs, once activated, further modulates ECM through various mechanisms. Previous studies have suggested that tissue stiffness can enhance the proliferation of tumor cells [63][104]. Collagen cross-linking by Lysyl-oxidase (LOX) and Tissue-transglutaminase 2 (TG2) enhances matrix stiffening [64][65][105,106]. Driven by TG2 and/or LOX, additional cross-linked collagen and rigid ECM activate Yes-associated protein (YAP) and the transcriptional coactivators of PDZ-binding motifs (TAZ), which enhances cell proliferation [66][107]. The stiffened ECM matrix in turn regulates the activity of ECM MMPs, Vimentin, and E-cadherin [66][67][107,108].3. ECM and Pancreatic Cancer Stem Cells (PCSCs)
The tumorigenic ability of each PDAC cell is heterogenous, and the development and proliferation of PDAC are highly dependent on the restricted PDAC cell subpopulation, namely pancreatic cancer stem cells (PCSCs) [68][109]. The activity of stem cells is dependent on exogenous niche factors in normal tissues and organs. Similarly, significant changes within the tumor microenvironment in PDAC can have a similar effect on PCSCs. The tumor microenvironment of PDAC strongly promotes fibroplasia, in which the ECM interacts with integrins (including integrin subunits β1, α6, and β3) to regulate PCSCs function by influencing autocrine and paracrine signaling pathways [69][110]. Type 1 collagen is the main scaffold for CD133-positive CSCs and promotes cell invasion through the PI3K/AKT pathway and also increases PCSCs enrichment by activating Focal Adhesion Kinase (FAK) [70][111]. Abnormal collagen cross-linking creates mechanical stresses that increase the rigidity and stiffness of the cancer matrix, which are important factors that are transmitted to CSC and regulate its proliferation and plasticity [71][112]. The ECM can also dynamically influence CSC ecology by creating a hypoxic environment [72][113]. Nevertheless, proteoglycans bind to various cytokines and chemokines in the tumor microenvironment to activate multiple signaling pathways in CSCs, including Notch, Wnt, and Hedgehog [73][117]. Transcriptional activation, mediated by the Wnt/β-catenin signaling pathway, induces the expression of C-MYC and SOX2, thereby promoting cancer stemness [74][118]. Glypican-4 (GPC4), as a typical proteoglycan, is associated with various human malignancies [75][76][119,120]. In addition, hyaluronic acid is a major ECM component of the stem cell niche, is frequently overexpressed in PDAC, and affects stromal cell behavior, which provides a favorable microenvironment for CSC self-renewal and maintenance [77][123]. This process mainly relies on the interaction of hyaluronic acid with its main receptor CD44. The hyaluronic acid-CD44 axis modulates the stemness properties of CSCs by inducing the EMT program and the secretion of extracellular vesicles. Specifically, excessive hyaluronic acid can activate AKT and ERK1/2 to induce EMT, and CD44 was found to be an important factor in TGF-β-induced EMT [78][124]. Intracellular hyaluronic acid-induced EMT increases centrosome abnormalities and micronucleation, creating a suitable niche for CSCs [79][125].4. ECM, PCSCs, and Metastasis
PCSCs, one of the main forces that are responsible for cancer metastasis, have the ability to evade treatment, exosmosis, and colonization from the primary tumor site to the site of secondary metastasis [80][128]. The niche formation of the metastatic site drives the transfer of CSCs [7]. Interactions between CSCs and the microenvironment such as CAFs and immune cells, as well as with the cell matrix, contribute to CSC migration and the priming of metastatic sites [7]. Another major factor in the formation of metastatic niches is the presence of extracellular vesicles, which are exosomes that also contain ECM regulatory genes [71][112]. The cross-linking of collagen and elastin in the ECM is primarily regulated by the LOX protein family, which consists of LOX-1 and four related enzymes, LOX -like protein (LOXL1-4) [81][82][83][129,130,131]. LOXL2, in particular, leads to the stiffening of PDAC tissues by promoting the cross-linking of collagen fibers, and increases the secretion of related factors (such as exosomes) in primary tumor tissues, which leads to ECM remodeling or stromal cell recruitment, and promotes the formation of an ecological niche before secondary organ metastasis, and ultimately promotes metastatic formation [84][132]. Furthermore, the overexpression of LOXL2 in mouse models increases EMT and stemness, thereby promoting primary and metastatic tumor growth and reducing the overall survival [84][132]. Some specific ECM molecules, including hyaluronic acid and Tenascin-C, have become the focus of research on the mechanism of tumor metastasis to specific sites [85][86][133,134]. CD44, a PCSC marker, has been identified as a Hyaluronic acid receptor that activates the EMT pathway and promotes pancreatic cancer metastasis [86][134]. Tenascin-C enhances the expression of stem cell signaling components musashi homolog 1 (MSI1) and leucine-rich repeat G protein-coupled receptor 5 (LGR5). These molecules are positive regulators of the Notch signaling and target genes of the Wnt pathway, respectively, and promote the formation and growth of metastases [85][133]. The ECM components and their biological and physical properties are involved in the regulation of basic functions of immune cells, such as activation, proliferation and migration. The previously described M2 macrophages engineered with CAFs can drive cancer cell invasion in a CCL18-dependent manner [87][135]. Cytotoxic T-lymphocytes (CTLs) physiologically play an important role in eliminating cancer cells, but studies have found that they are often trapped in dense ECM compartments. Immune cells that are initially attracted to the tumor site by chemokines are prevented from migrating to the tumor core following contact with the area of increased stiffness [88][89][66,136]. Continuous ECM remodeling and overexpression of certain matrix components also promoted the recruitment of bone marrow cells, which are ultimately polarized to support ECM remodeling, CTL inhibition, tumor proliferation, and invasion [90][91][137,138]. The process of CSC migration involves ECM degradation, remodeling of cell–cell and cell–ECM interactions, formation of adhesive plaques and invasiveness, and EMT transformation, which requires a lot of cellular energy [92][144]. The glycolysis dependence of CSCs can drive the degradation of the ECM, the formation of invasive structures, and cellular protrusions, leading to the migration and invasion of cancer cells [93][145]. The YAP/TAZ transcriptional coactivator is a core component of the Hippo pathway and a sensor of the structural and mechanical characteristics of the cellular microenvironment [94][153]. Increased matrix stiffness in PDAC leads to YAP/TAZ activation, which in turn promotes the production of profibrotic mediators and ECM proteins. Activated YAP/TAZ can enhance tumor proliferation and survival by transactivating target genes that are related to cell cycle progression and anti-apoptosis and can also induce the expression of the proto-oncogene c-Myc to promote cell cycle progression [95][96][154,155]. More importantly, YAP/TAZ is closely related to ZEB1/2 and Twist, both of which can be used to regulate EMT, thereby inducing malignant features of cancer cells and CSC-related properties, such as tumor initiation, drug resistance, and metastasis [97][156].56. ECM, PCSCs, and Chemoresistance
To date, the mechanisms of ECM-related chemoresistance have been identified, which can be roughly divided into two major categories, namely physical barriers (abnormal vascularization and matrix stiffness) and cell adhesion-related means (ECM tissue, mechanical signaling pathways, and pro-survival signaling pathways) [98][158] (Figure 1). Aberrant ECM remodeling results in increased matrix stiffness, vessel collapse, and reduced blood flow, which greatly reduces the ability of drugs to enter the tumor [99][159]. As previously described, dense fibrosis and abnormal vascularization in PDAC contribute to the formation of a hypoxic and pH-abnormal tumor microenvironment. Hypoxia also affects drug movement from the bloodstream to the tumor microenvironment, specifically affecting the activity of drug transporters and the expression and activity of phase I drug metabolizing enzymes [100][101][102][160,161,162]. Glycolysis under hypoxia results in the production of large amounts of lactic acid, which reduces the extracellular pH. The ability of the drug to cross the hydrophobic membrane is greatly reduced when the drug is electrically charged in an acidic environment [100][160]. In addition to the dense fibrotic ECM that impairs the ability of drugs to spread from blood vessels to cancer cells, most ECM proteins contribute to chemoresistance by activating EMT and oncogenic signaling pathways, including MAPK, PI3K, and YAP [103][104][105][106][163,164,165,166]. CSCs are also an important factor leading to chemotherapy resistance, but there are only few studies that are available on the relationship between the ECM and PCSC in chemoresistance. Therefore, this part focuses on the research progress of ECM-PCSC interactions and chemoresistance.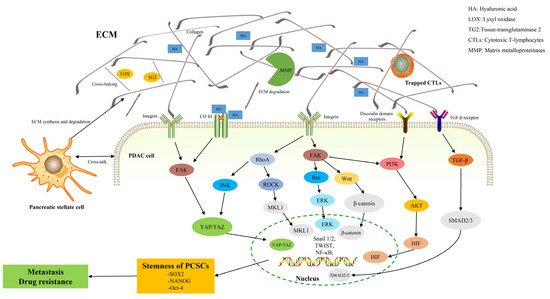
6. Pharmacological Targeting of ECM
Collagen is one of the most basic components in the ECM, and it is also one of the ideal therapeutic directions. TGF-β plays a key role in collagen synthesis, so TGF-β signaling is the most promising target for inhibiting collagen synthesis. In animal models of PDAC, an anticoccidial named halofuginone has been shown to reduce collagen synthesis by inhibiting TGF-β signaling [112][181]. TGF-β is usually overexpressed in PDAC and blocking TGF-β-mediated signaling may enhance antitumor effects [110][111][173,174]. A clinical trial aiming to target TGF-β in PDAC is investigating the antitumor activity of SAR439459, a pan-TGF-β neutralizing antibody [113][182]. Vitamin D may also block collagen secretion by disrupting the TGF-β signaling pathway, helping to prevent tumor metastasis and enhance drug responses [114][115][183,184]. The inhibition of collagen cross-linking is also a therapeutic strategy to target ECM stiffness in cancer. In 2017, a randomized Phase II study of Simtuzumab, a humanized IgG4 monoclonal antibody that inhibits extracellular LOXL2, was performed in combination with gemcitabine in patients that were suffering from metastatic PDAC [84][132]. However, the results showed that a combination of gemcitabine and Simtuzumab did not improve clinical outcomes. Therefore, the complex tumor-stromal interaction of LOXL2 in PDAC tumorigenesis requires further investigation and re-evaluation of the efficacy of using anti-LOXL2 antibodies or small molecule LOXL2 inhibitors in the treatment landscape of metastatic PDAC [116][185].
There are other therapeutic ideas aiming to target the MMP to alter ECM stiffness. For example, MMP-9 promotes aggressive and metastatic phenotypes in tumor cells, and its overexpression increases the aggressiveness of cancer cell lines in vitro [117][194]. Targeting integrin-mediated signaling is another strategy for ECM therapy. In theory, it is possible to disrupt signals from the extracellular or intracellular environment by using integrin inhibitors to disrupt ECM mechanical sensing [118][177]. Targeted glutamine metabolism and hexosamine biosynthesis pathways has been shown to have profound effects on the ECM and CSC self-renewal of PDAC among metabolic inhibitors [119][195].
Clinical trials involving different inhibitors are being conducted in different clinical settings, primarily for ECM-induced stemness of PCSCs. For example, the inhibitors of Rho-associated kinase (ROCK) alter the contractility of PDAC cytoskeleton and CAF, may benefit drug delivery and inhibit metastasis. ECM disorder occurrence and PDAC migration and invasion are prevented when ROCK is suppressed. Other mouse models showed that a pre-chemotherapy administration of ROCK inhibitors increased the chemotherapy response of the primary tumor and helped to prevent the development of liver metastases [120][121][122][202,203,204]. T13148 was the first ROCK inhibitor that was investigated for the treatment of solid tumors, but further development of this compound is advised due to drug side effects [123][205].