 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Henry Fechner | -- | 2005 | 2022-08-25 11:00:02 | | | |
| 2 | Camila Xu | + 4 word(s) | 2009 | 2022-08-26 02:22:27 | | |
Video Upload Options
Coxsackievirus B3 (CVB3) is a non-enveloped single-stranded RNA virus belonging to the genus Enterovirus of the picornavirus family.
1. Coxsackievirus B3 (CVB3) Structure, Genome and Protein Functions
CVB3 is a non-enveloped single-stranded RNA virus belonging to the genus Enterovirus of the picornavirus family. As with all members of the picornaviridae, CVB3 is characterized by an icosahedral capsid of approximately 30 nm diameter, which houses the positive-sense (+) RNA genome [1][2]. The capsid consists of twelve pentamers, each composed of five asymmetric units of the structural proteins VP1–VP4 (Figure 1A). VP1 to VP3 form the viral shell. VP4 lies at the inner surface of the viral shell making a connection between N-termini of the other capsid proteins and the viral RNA, thereby acting as a stabilizer of the capsid pentamers during virus assembly [3][4].

The capsid surface forms a depression, called the canyon, around the five-fold axis of symmetry of each pentamer [5] (Figure 1B). Underneath the bottom of the canyon there is a hydrophobic pocket hosting a C16 fatty acid which is referred to as the pocket factor and contributes to the stability of the viral capsid [6][7][8]. It is thought that the binding of the Coxsackievirus and Adenovirus Receptor (CAR) [9][10] to the pocket displaces the pocket factor, thereby destabilizing the capsid, triggering the uncoating and delivery of the viral RNA into the cells [7][8][11]. Another important structural feature of the capsid surface, the elevated hypervariable puff region, located at the southern rim of the canyon (Figure 1B), functions as a known antigenic site [5][12][13]. Furthermore, it is involved in the binding of the decay accelerating factor (DAF) which serves as a co-receptor of CVB3 [14][15].
The positive-sense (+) RNA genome of CVB3 has a length of approximately 7.5 kilobase pairs (kb). It comprises a single large open reading frame (ORF) flanked by a 742 nucleotide (nt) long 5′-untranslated region (5′-UTR) and an about 100 nt long polyadenylated 3‘-UTR [1]. Particularly the long 5′-UTR builds a number of stem-loop structures, among them the cloverleaf (CL) and the internal ribosomal entry site (IRES) which play major roles in viral replication and protein synthesis [1][16][17][18][19]. The CL interacts with VPg (virus protein genome-linked, also known as 3B), which is covalently attached to the 5′-end of the positive-sense RNA, and with the 3′-UTR and other trans-acting proteins to form the replication complex during RNA synthesis [18][20][21][22][23][24]. The IRES conveys the CAP-independent interaction with the cellular ribosome for viral translation [17][20]. The ORF encodes a continuous polyprotein which is autocatalytically processed into the 4 structural (VP–VP4) and 7 non-structural proteins (2A–2C, 3A–3D), as well as 3 intermediate cleavage products (2BC, 3AB and 3CD) [20] (Figure 2).
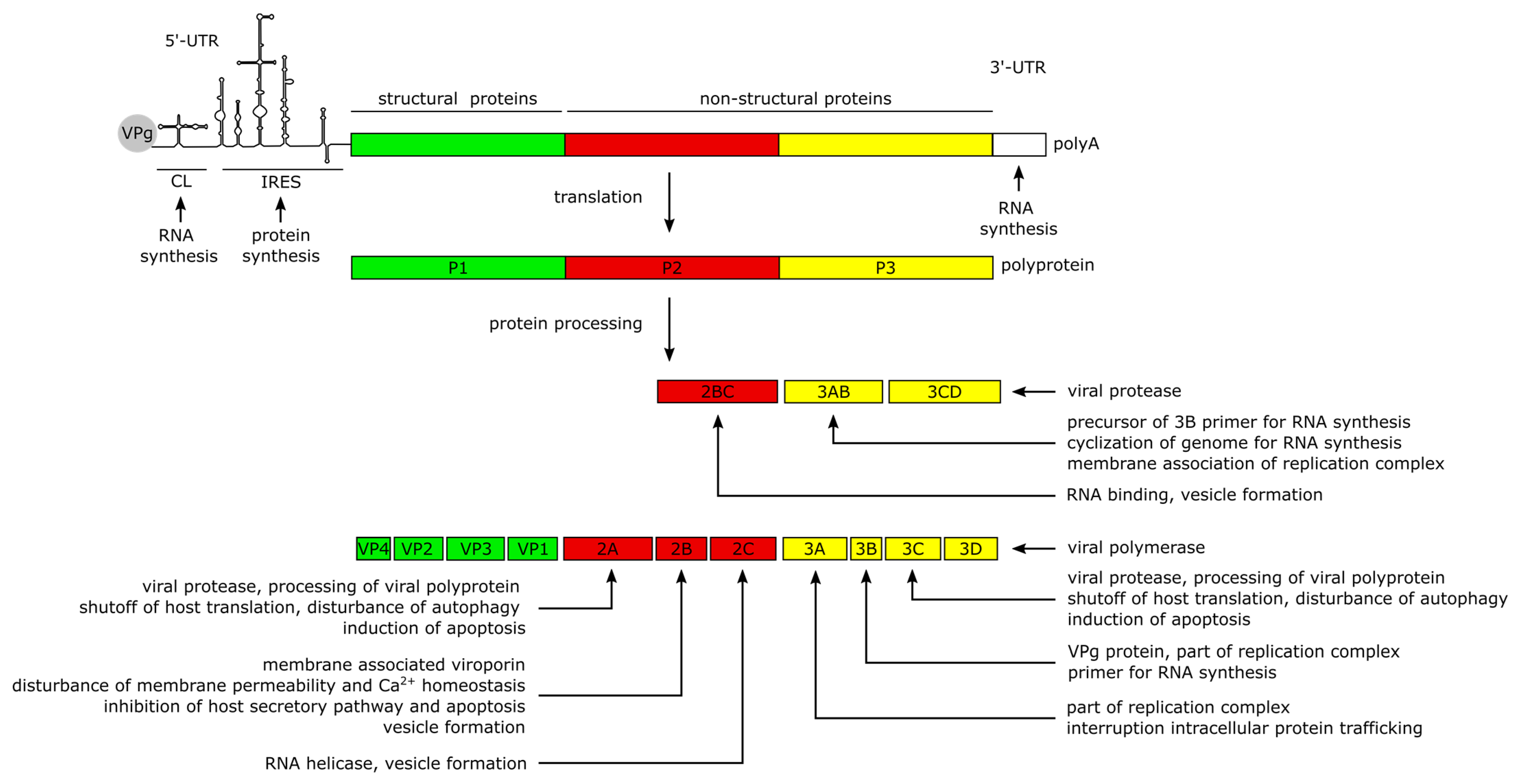
The non-structural proteins function to promote viral protein synthesis, replication, release and spread by interacting with the RNA genome and polyproteins, while also interfering with cellular processes. Most of the manipulation of host cell processes and virus-induced pathogenesis can be traced to the activities of viral proteases 2A and 3C. Besides the proteolytic processing of the polyprotein into the 11 structural and non-structural proteins, the proteases are involved in the shutdown of host cell translation and transcription, disruption of the cytoskeleton, induction of apoptosis and attenuation of the innate immune response. The blockage of translation is mainly carried out by cleavage of host factors like the eukaryotic initiation factor 4G [25], the poly(A)-binding protein [26] and the Death-Associated Protein 5, as they are important mediators of cap-dependent and IRES-dependent translation initiation in the cell [1][27][28]. To prevent premature viral clearance from the cell, the proteases also cleave the immune adaptor molecules and pro-apoptotic factors, named Mitochondrial Antiviral Signaling Protein (MAVS) and Toll/IL-1 Receptor Domain-containing Adaptor Inducing Interferon-β (TRIF), which leads to an attenuated type I interferon response and apoptotic signaling during the early stages of CVB3 infection [29]. In addition, Protease 2A cleaves the cytoskeletal protein Dystrophin, an event shown to be important for the pathogenesis of CV-induced cardiomyopathies [30]. Another key feature of the 2A and 3C proteases is their ability to induce apoptosis through caspase-8-mediated activation of caspase-3 and to activate the intrinsic mitochondria-mediated apoptosis pathway during the late phase of viral infection [25].
The Viroporin 2B and its precursor 2BC build homo- and heteromultimers, which integrate into the membranes of the Golgi apparatus and the endoplasmic reticulum (ER) [31][32][33]. The resulting pore formation leads to a leakage of Ca2+ into the cytoplasm [31][32][34], disturbing pro-apoptotic signaling during the early stages of infection [32][35][36] thereby preventing a rapid clearance of the virus. Furthermore, the membrane interaction of 2B is thought to induce the formation of vesicles which are important for viral replication and release [34][35][37]. In addition, the 2C protein possesses a RNA helicase function in enteroviruses [38] which could also be confirmed for CVB3 [39][40].
The proteins 3A, 3AB and 3D interact with the viral genome to form the replication complex [21]. The 3B protein serves as the primer for the transcription of the viral genome [41]. The binding of 3AB is thought to activate the protease activity of CL-bound 3CD precursor to release the 3D polymerase and mediate the cyclization of the genome by interacting with the 3′-UTR during (−) RNA synthesis [22][42][43].
2. CVB3 Infections in Humans and in Experimentally Infected Mice
3. CVB3 Receptors and Its Importance for CVB3 Targeting of Cancer
Occurrence of viral receptors on the cell surface is a key feature that contributes to virus tropism. Hence, the expression of CVB3 receptors on cancer cells is vital for the successful treatment of cancer with oncolytic CVB3. The main receptor for CVB3 binding and uptake is CAR [10][60][61] (Figure 3 and Figure 4), a transmembrane protein which is involved in cell adhesion and inflammation [62]. In addition to CAR, several CVB3 strains, such as RD and HA, use DAF, which is involved in the regulation of complement activation and cell signaling. DAF functions as co-receptor for CVB3 attachment to the host cell surface [63][64]. The binding of DAF alone, however, is not sufficient to mediate viral entry into the cell and subsequent lytic infection [8][64]. Thus, cancer cells that express DAF but not CAR are not vulnerable to oncolytic CVB3.

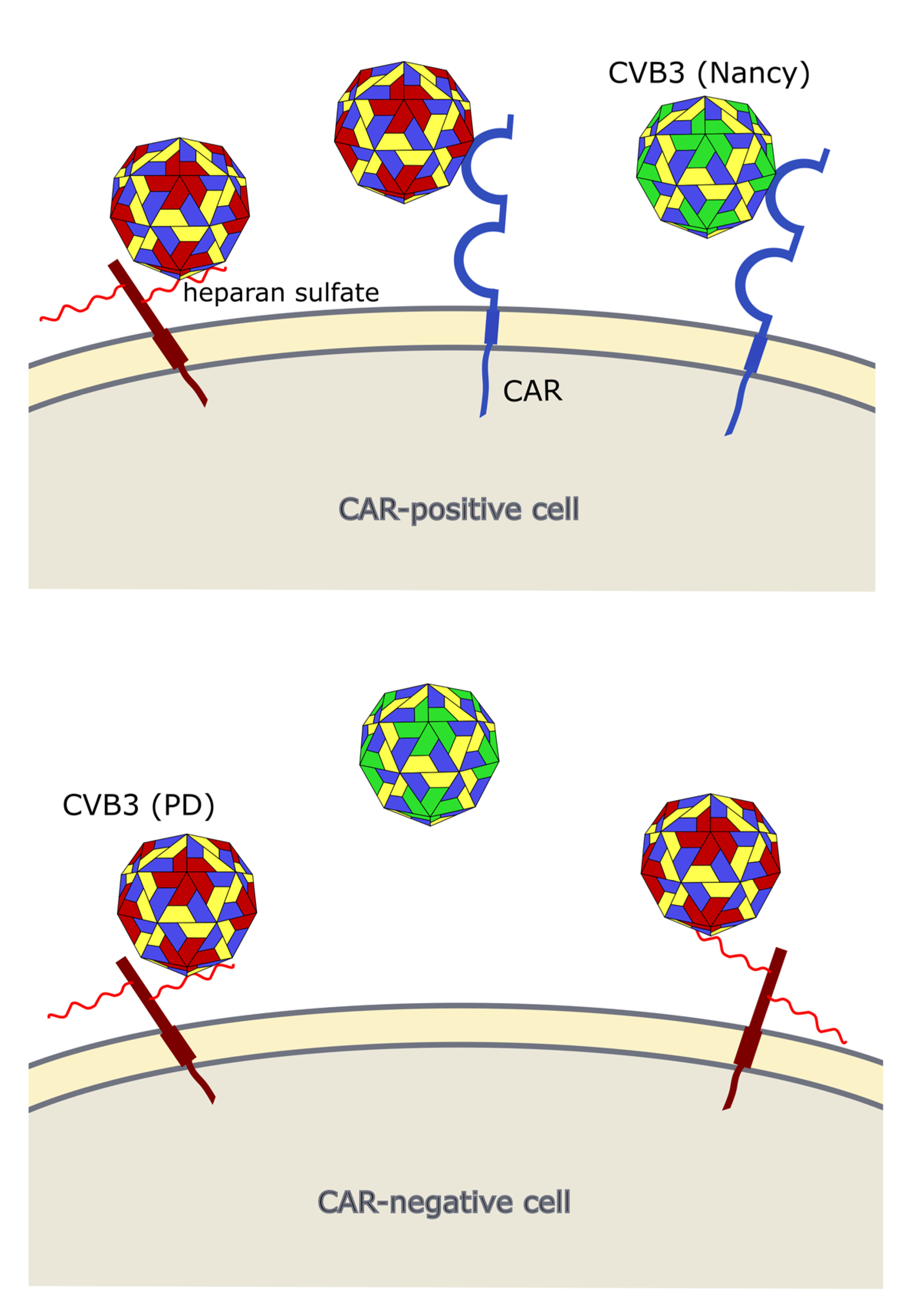
CAR is expressed in many tissues, including heart, lung, liver, testis, pancreas and kidney [66][67]. It is highly expressed during fetal development and in young individuals, while it is downregulated in adults [68]. In cancer, CAR is differentially expressed. Compared to normal tissues in lung cancer, cervical cancer, endometrial cancer, ovarian cancer and urinary bladder cancer, for example, CAR appears to be upregulated, whereas in colon and prostate cancers, as well as subtypes of renal cell cancers it is strongly downregulated [69].
Two studies, investigating lung [70] and endometrial cancer [71], found a good correlation between sensitivity of the cancer cell line to oncolytic CVB3 and their CAR and DAF expression levels. In another study, however, there was no clear correlation between abundance of CAR and susceptibility of colorectal carcinoma cell lines to oncolytic CVB3 [72], which may mean that under certain conditions post-entry mechanisms may be of particular importance for cytolytic activity of oncolytic CVB3.
In addition to CAR, it has been shown that CVB3 can use heparan sulfates to enter cancer cells. Thus far, however, this has only been shown for the CVB3 variant PD, which uses N- and 6-O-sulfated heparan sulfates to infect cells [72][73][74] (Figure 3 and Figure 4). Heparan sulfates are linear polysaccharides, which consist of repeating disaccharides bound to a core protein which links them to the cell surface. Based on the analysis of the expression of the heparan sulfate D-glucosaminyl 6-O-sulfotransferase-2 (HS6ST2), which catalyzes the transfer of sulfate groups to the C-6 (exocyclic carbon) of the glucosamine residue in heparan sulfate proteoglycans, the stomach, liver, adrenal gland, bronchus, breast, ovary, uterus, kidney and skin contain N- and 6-O-sulfated heparan sulfates. In other organs, such as the lung, pancreas, heart, spleen, prostate and colon, HS6STS expression could not be detected [75]. HS6ST2 is also differentially expressed in cancer. This enzyme is downregulated in ovarian cancer [76] but overexpressed in colorectal, gastric and pancreatic cancer [75][77][78].
A recent study from researcher group confirmed the importance of N- and 6-O-sulfated heparan sulfates for infection of cancer cells with the CVB3 variant PD. In fact, there was a positive correlation between expression HS6ST2 and the sensitivity of colorectal cancer cell lines to the PD strain of CVB3 [72].
References
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82.
- Ramsingh, A.I.; Collins, D.N. A point mutation in the VP4 coding sequence of coxsackievirus B4 influences virulence. J. Virol. 1995, 69, 7278–7281.
- Ansardi, D.C.; Porter, D.C.; Morrow, C.D. Myristylation of poliovirus capsid precursor P1 is required for assembly of subviral particles. J. Virol. 1992, 66, 4556–4563.
- Organtini, L.J.; Makhov, A.M.; Conway, J.F.; Hafenstein, S.; Carson, S.D. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J. Virol. 2014, 88, 5755–5765.
- Muckelbauer, J.K.; Kremer, M.; Minor, I.; Diana, G.; Dutko, F.J.; Groarke, J.; Pevear, D.C.; Rossmann, M.G. The structure of coxsackievirus B3 at 3.5 å resolution. Structure 1995, 3, 653–667.
- Oliveira, M.A.; Zhao, R.; Lee, W.-M.; Kremer, M.J.; Minor, I.; Rueckert, R.R.; Diana, G.D.; Pevear, D.C.; Dutko, F.J.; A McKinlay, M.; et al. The structure of human rhinovirus 16. Structure 1993, 1, 51–68.
- Milestone, A.M.; Petrella, J.E.; Sanchez, M.D.; Mahmud, M.; Whitbeck, J.C.; Bergelson, J.M. Interaction with coxsackievirus and adenovirus receptor, but not with decay-accelerating factor (DAF), induces A-particle formation in a DAF-binding coxsackievirus B3 isolate. J. Virol. 2005, 79, 655–660.
- Bergelson, J.M.; Krithivas, A.; Celi, L.; Droguett, G.; Horwitz, M.S.; Wickham, T.; Crowell, R.L.; Finberg, R.W. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J. Virol. 1998, 72, 415–419.
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356.
- Rossmann, M.G. Viral cell recognition and entry. Protein Sci. 1994, 3, 1712–1725.
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nat. Cell Biol. 1985, 317, 145–153.
- Hogle, J.M.; Chow, M.; Filman, D.J. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 1985, 229, 1358–1365.
- Hafenstein, S.; Bowman, V.D.; Chipman, P.R.; Kelly, C.M.B.; Lin, F.; Medof, M.E.; Rossmann, M.G. Interaction of decay-accelerating factor with coxsackievirus B3. J. Virol. 2007, 81, 12927–12935.
- Yoder, J.D.; Cifuente, J.O.; Pan, J.; Bergelson, J.M.; Hafenstein, S. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) Provide a new footprint of DAF on the virus surface. J. Virol. 2012, 86, 12571–12581.
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380.
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nat. Cell Biol. 1988, 334, 320–325.
- Zell, R.; Sidigi, K.; Bucci, E.; Stelzner, A.; Görlach, M. Determinants of the recognition of enteroviral cloverleaf RNA by cox-sackievirus B3 proteinase 3C. RNA 2002, 8, 188–201.
- Bailey, J.M.; Tapprich, W.E. Structure of the 5′ nontranslated region of the coxsackievirus B3 genome: Chemical modification and comparative sequence analysis. J. Virol. 2006, 81, 650–668.
- Lin, J.-Y.; Chen, T.-C.; Weng, K.-F.; Chang, S.-C.; Chen, L.-L.; Shih, S.-R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16, 103.
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A kidnapping story: How coxsackievirus B3 and its host cell interact. Cell. Physiol. Biochem. 2019, 53, 121–140.
- Xiang, W.; Harris, K.S.; Alexander, L.; Wimmer, E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of po-liovirus is essential for RNA replication. J. Virol. 1995, 69, 3658–3667.
- Andino, R.; Rieckhof, G.; Achacoso, P.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993, 12, 3587–3598.
- Melchers, W.J.; Hoenderop, J.G.; Slot, H.J.B.; Pleij, C.W.; Pilipenko, E.V.; I Agol, V.; Galama, J.M. Kissing of the two predominant hairpin loops in the coxsackie B virus 3’ untranslated region is the essential structural feature of the origin of replication required for negative-strand RNA synthesis. J. Virol. 1997, 71, 686–696.
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2006, 12, 513–524.
- Kerekatte, V.; Keiper, B.D.; Badorff, C.; Cai, A.; Knowlton, K.U.; Rhoads, R.E. Cleavage of poly(A)-binding protein by cox-sackievirus 2A protease in vitro and in vivo: Another mechanism for host protein synthesis shutoff? J. Virol. 1999, 73, 709–717.
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eu-karyotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113.
- Hanson, P.J.; Ye, X.; Qiu, Y.; Zhang, H.M.; Hemida, M.G.; Wang, F.; Lim, T.; Gu, A.; Cho, B.; Kim, H.; et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 2015, 23, 828–840.
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311.
- Lim, B.-K.; Peter, A.K.; Xiong, D.; Narezkina, A.; Yung, A.; Dalton, N.D.; Hwang, K.-K.; Yajima, T.; Chen, J.; Knowlton, K.U. Inhibition of Coxsackievirus-associated dystrophin cleavage prevents cardiomyopathy. J. Clin. Investig. 2013, 123, 5146–5151.
- de Jong, A.S.; Wessels, E.; Dijkman, H.B.P.M.; Galama, J.M.D.; Melchers, W.J.G.; Willems, P.H.G.M.; van Kuppeveld, F.J.M. Determinants for membrane association and permeabilization of the coxsackievirus 2B protein and the identification of the golgi complex as the target organelle. J. Biol. Chem. 2003, 278, 1012–1021.
- de Jong, A.S.; Visch, H.-J.; de Mattia, F.; van Dommelen, M.M.; Swarts, H.G.; Luyten, T.; Callewaert, G.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. The coxsackievirus 2B protein increases efflux of ions from the endoplasmic reticulum and golgi, thereby inhibiting protein trafficking through the golgi. J. Biol. Chem. 2006, 281, 14144–14150.
- De Jong, A.S.; Schrama, I.W.J.; Willems, P.H.G.M.; Galama, J.M.D.; Melchers, W.J.G.; Van Kuppeveld, F.J.M. Multimerization reactions of coxsackievirus proteins 2B, 2C and 2BC: A mammalian two-hybrid analysis. J. Gen. Virol. 2002, 83, 783–793.
- Van Kuppeveld, F.J.; Hoenderop, J.G.; Smeets, R.L.; Willems, P.H.; Dijkman, H.B.; Galama, J.M.; Melchers, W.J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997, 16, 3519–3532.
- Buenz, E.J.; Howe, C.L. Picornaviruses and cell death. Trends Microbiol. 2006, 14, 28–36.
- Campanella, M.; de Jong, A.S.; Lanke, K.W.H.; Melchers, W.J.G.; Willems, P.H.G.M.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J.M. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450.
- Van Kuppeveld, F.J.; Galama, J.M.; Zoll, J.; Hurk, P.J.V.D.; Melchers, W.J. Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J. Virol. 1996, 70, 3876–3886.
- Xia, H.; Wang, P.; Wang, G.C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; Shu, T.; Zhao, X.; Yin, L.; et al. Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLoS Pathog. 2015, 11, e1005067.
- Fang, Y.; Wang, C.; Yang, R.; Bai, P.; Zhang, X.-Y.; Kong, J.; Yin, L.; Qiu, Y.; Zhou, X. Antiviral peptides targeting the helicase activity of enterovirus nonstructural protein 2C. J. Virol. 2021.
- Yun, S.-H.; Shin, H.-H.; Ju, E.-S.; Lee, Y.-J.; Lim, B.-K.; Jeon, E.-S. Inhibition of RNA helicase activity prevents coxsackievirus B3-induced myocarditis in human iPS cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 3041.
- Paul, A.V.; van Boom, J.H.; Filippov, D.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 1998, 393, 280–284.
- Molla, A.; Harris, K.S.; Paul, A.V.; Shin, S.H.; Mugavero, J.; Wimmer, E. Stimulation of poliovirus proteinase 3Cpro-related proteolysis by the genome-linked protein VPg and its precursor 3AB. J. Biol. Chem. 1994, 269, 27015–27020.
- Harris, K.S.; Xiang, W.; Alexander, L.; Lane, W.S.; Paul, A.V.; Wimmer, E. Interaction of poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 1994, 269, 27004–27014.
- Bowles, N.E.; Richardson, P.J.; Olsen, E.G.; Archard, L.C. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1986, 1, 1120–1123.
- Compagnoli Carmona, R.D.; Caetano Machado, B.; Aparecida de Sousa, C.; Vieira, H.R.; Moraes Alves, M.R.; Farias de Souza, K.A.; de Souza Gregório, D.; Costa Vilanova, B.; Sampaio Tavares Timenetsky, M.D. Distribution of species enterovirus B in patients with central nervous system infections in São Paulo State, Brazil. J. Med. Virol. 2020, 92, 3849–3856.
- Foulis, A.K.; Farquharson, M.A.; Cameron, S.O.; McGill, M.; Schonke, H.; Kandolf, R. A search for the presence of the enteroviral capsid protein VP1 in pancreases of patients with type 1 (insulin-dependent) diabetes and pancreases and hearts of infants who died of coxsackieviral myocarditis. Diabetologia 1990, 33, 290–298.
- Tracy, S.; Hofling, K.; Pirruccello, S.; Lane, P.H.; Reyna, S.M.; Gauntt, C.J. Group B coxsackievirus myocarditis and pancreatitis: Connection between viral virulence phenotypes in mice. J. Med. Virol. 2000, 62, 70–81.
- Wang, S.-M.; Liu, C.-C.; Yang, Y.-J.; Yang, H.-B.; Lin, C.-H.; Wang, J.-R. Fatao coxsackievirus B infection in early infancy characterized by fulminant hepatitis. J. Infect. 1998, 37, 270–273.
- Tariq, N.; Kyriakopoulos, C. Group B Coxsackie Virus; StatPearls: Treasure Island, FL, USA, 2021.
- Iwasaki, T.; Monma, N.; Satodate, R.; Kawana, R.; Kurata, T. An immunofluorescent study of generalized Coxsackie virus B3 infection in a newborn infant. Acta Pathol. Jpn. 1985, 35, 741–748.
- Ronellenfitsch, S.; Tabatabai, J.; Bottcher, S.; Diedrich, S.; Frommhold, D.; Schubert-Bast, S.; Poeschl, J.; Schnitzler, P. First report of a Chinese strain of coxsackie B3 virus infection in a newborn in Germany in 2011: A case report. J. Med. Case Rep. 2014, 8, 164.
- Liu, J.-Y.; Wang, S.-M.; Chen, I.-C.; Yu, C.-K.; Liu, C.-C. Hepatic damage caused by coxsackievirus B3 is dependent on age-related tissue tropisms associated with the coxsackievirus-adenovirus receptor. Pathog. Dis. 2013, 68, 52–60.
- Pinkert, S.; Pryshliak, M.; Pappritz, K.; Knoch, K.; Hazini, A.; Dieringer, B.; Schaar, K.; Dong, F.; Hinze, L.; Lin, J.; et al. Development of a new mouse model for coxsackievirus-induced myocarditis by attenuating coxsackievirus B3 virulence in the pancreas. Cardiovasc. Res. 2020, 116, 1756–1766.
- Kallewaard, N.L.; Zhang, L.; Chen, J.W.; Guttenberg, M.; Sanchez, M.D.; Bergelson, J.M. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 2009, 6, 91–98.
- Pinkert, S.; Westermann, D.; Wang, X.; Klingel, K.; Dorner, A.; Savvatis, K.; Grossl, T.; Krohn, S.; Tschope, C.; Zeichhardt, H.; et al. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 2009, 120, 2358–2366.
- Schmidtke, M.; Merkle, I.; Klingel, K.; Hammerschmidt, E.; Zautner, A.E.; Wutzler, P. The viral genetic background determines the outcome of coxsackievirus B3 infection in outbred NMRI mice. J. Med. Virol. 2007, 79, 1334–1342.
- Koenig, A.; Buskiewicz, I.; Huber, S.A. Age-associated changes in estrogen receptor ratios correlate with increased female susceptibility to coxsackievirus B3-induced myocarditis. Front. Immunol. 2017, 8, 1585.
- Li, K.; Xu, W.; Guo, Q.; Jiang, Z.; Wang, P.; Yue, Y.; Xiong, S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009, 105, 353–364.
- Leipner, C.; Grun, K.; Schneider, I.; Gluck, B.; Sigusch, H.H.; Stelzner, A. Coxsackievirus B3-induced myocarditis: Differences in the immune response of C57BL/6 and Balb/c mice. Med. Microbiol. Immunol. 2004, 193, 141–147.
- Carson, S.D.; Chapman, N.N.; Tracy, S.M. Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem. Biophys. Res. Commun. 1997, 233, 325–328.
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323.
- Ortiz-Zapater, E.; Santis, G.; Parsons, M. CAR: A key regulator of adhesion and inflammation. Int. J. Biochem. Cell Biol. 2017, 89, 1–5.
- Bergelson, J.M.; Mohanty, J.G.; Crowell, R.L.; St John, N.F.; Lublin, D.M.; Finberg, R.W. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J. Virol. 1995, 69, 1903–1906.
- Shafren, D.R.; Bates, R.C.; Agrez, M.V.; Herd, R.L.; Burns, G.F.; Barry, R.D. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 1995, 69, 3873–3877.
- He, Y.; Chipman, P.R.; Howitt, J.; Bator, C.M.; Whitt, M.A.; Baker, T.S.; Kuhn, R.J.; Anderson, C.W.; Freimuth, P.; Rossmann, M.G. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Genet. 2001, 8, 874–878.
- Fechner, H.; Haack, A.; Wang, H.; Wang, X.; Eizema, K.; Pauschinger, M.; Schoemaker, R.; Veghel, R.; Houtsmuller, A.; Schultheiss, H.P.; et al. Expression of coxsackie adenovirus receptor and alphav-integrin does not correlate with adenovector targeting in vivo indicating anatomical vector barriers. Gene Ther. 1999, 6, 1520–1535.
- Zanone, M.M.; Favaro, E.; Ferioli, E.; Huang, G.C.; Klein, N.J.; Perin, P.C.; Peakman, M.; Conaldi, P.G.; Camussi, G. Human pancreatic islet endothelial cells express coxsackievirus and adenovirus receptor and are activated by coxsackie B virus infection. FASEB J. 2007, 21, 3308–3317.
- Fechner, H.; Noutsias, M.; Tschoepe, C.; Hinze, K.; Wang, X.; Escher, F.; Pauschinger, M.; Dekkers, D.; Vetter, R.; Paul, M.; et al. Induction of coxsackievirus-adenovirus-receptor expression during myocardial tissue formation and remodeling: Identification of a cell-to-cell contact-dependent regulatory mechanism. Circulation 2003, 107, 876–882.
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the coxsackievirus and adenovirus receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858.
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012, 72, 2609–2621.
- Lin, Y.; Wang, W.; Wan, J.; Yang, Y.; Fu, W.; Pan, D.; Cai, L.; Cheng, T.; Huang, X.; Wang, Y. Oncolytic activity of a coxsackievirus B3 strain in human endometrial cancer cell lines. Virol. J. 2018, 15, 65.
- Hazini, A.; Pryshliak, M.; Bruckner, V.; Klingel, K.; Sauter, M.; Pinkert, S.; Kurreck, J.; Fechner, H. Heparan sulfate binding coxsackievirus B3 strain PD: A novel avirulent oncolytic agent against human colorectal carcinoma. Hum. Gene Ther. 2018, 29, 1301–1314.
- Zautner, A.E.; Korner, U.; Henke, A.; Badorff, C.; Schmidtke, M. Heparan sulfates and coxsackievirus-adenovirus receptor: Each one mediates coxsackievirus B3 PD infection. J. Virol. 2003, 77, 10071–10077.
- Zautner, A.E.; Jahn, B.; Hammerschmidt, E.; Wutzler, P.; Schmidtke, M. N- and 6-O-sulfated heparan sulfates mediate internalization of coxsackievirus B3 variant PD into CHO-K1 cells. J. Virol. 2006, 80, 6629–6636.
- Hatabe, S.; Kimura, H.; Arao, T.; Kato, H.; Hayashi, H.; Nagai, T.; Matsumoto, K.; de Velasco, M.; Fujita, Y.; Yamanouchi, G.; et al. Overexpression of heparan sulfate 6-O-sulfotransferase-2 in colorectal cancer. Mol. Clin. Oncol. 2013, 1, 845–850.
- Cole, C.L.; Rushton, G.; Jayson, G.C.; Avizienyte, E. Ovarian cancer cell heparan sulfate 6-O-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (EGF)-like growth factor/EGF receptor signaling. J. Biol. Chem. 2014, 289, 10488–10501.
- Jin, Y.; He, J.; Du, J.; Zhang, R.-X.; Yao, H.-B.; Shao, Q.-S. Overexpression of HS6ST2 is associated with poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 14, 6191–6197.
- Song, K.; Li, Q.; Peng, Y.-B.; Li, J.; Ding, K.; Chen, L.-J.; Shao, C.-H.; Zhang, L.-J.; Li, P. Silencing of hHS6ST2 inhibits progression of pancreatic cancer through inhibition of Notch signalling. Biochem. J. 2011, 436, 271–282.

