 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Massimiliano Mazza | -- | 4148 | 2022-08-23 17:16:05 | | | |
| 2 | Vivi Li | -8 word(s) | 4140 | 2022-08-24 04:28:51 | | |
Video Upload Options
The immune system is a dynamic feature of each individual and a footprint of our unique internal and external exposures. Indeed, the type and level of exposure to physical and biological agents shape the development and behavior of this complex and diffuse system. Many pathological conditions depend on how our immune system responds or does not respond to a pathogen or a disease or on how the regulation of immunity is altered by the disease itself. T-cells are important players in adaptive immunity and, together with B-cells, define specificity and monitor the internal and external signals that our organism perceives through its specific receptors, TCRs and BCRs, respectively. Today, high-throughput sequencing (HTS) applied to the TCR repertoire has opened a window of opportunity to disclose T-cell repertoire development and behavior down to the clonal level. Although TCR repertoire sequencing is easily accessible today, it is important to deeply understand the available technologies for choosing the best fit for the specific experimental needs and questions. Here, an updated overview of TCR repertoire sequencing strategies, providers and applications to infectious diseases and cancer to guide researchers’ choice through the multitude of available options is provided. The possibility of extending the TCR repertoire to HLA characterization will be of pivotal importance in the near future to understand how specific HLA genes shape T-cell responses in different pathological contexts and will add a level of comprehension that was unthinkable just a few years ago.
1. Introduction
| Sources of α/β TCR Diversity | |
|---|---|
| Recombination of the T-cell α-genes on chromosome 14 and T-cell β-genes on chromosome 7 by RAG1/2 enzymes. | Total α-genes combination: 2392 Total β-genes combination: 1248 Total αβ-genes combination: 2,985,216 [6]. |
| Theoretical diversity by pairing of different in-frame α- and β-chains plus junctional diversity by terminal deoxynucleotidyl transferase activity [6]. | 1015–1061 [14] |
| Experimental diversity evaluation by deep sequencing. | 104–106, based on the amount of the sample. |
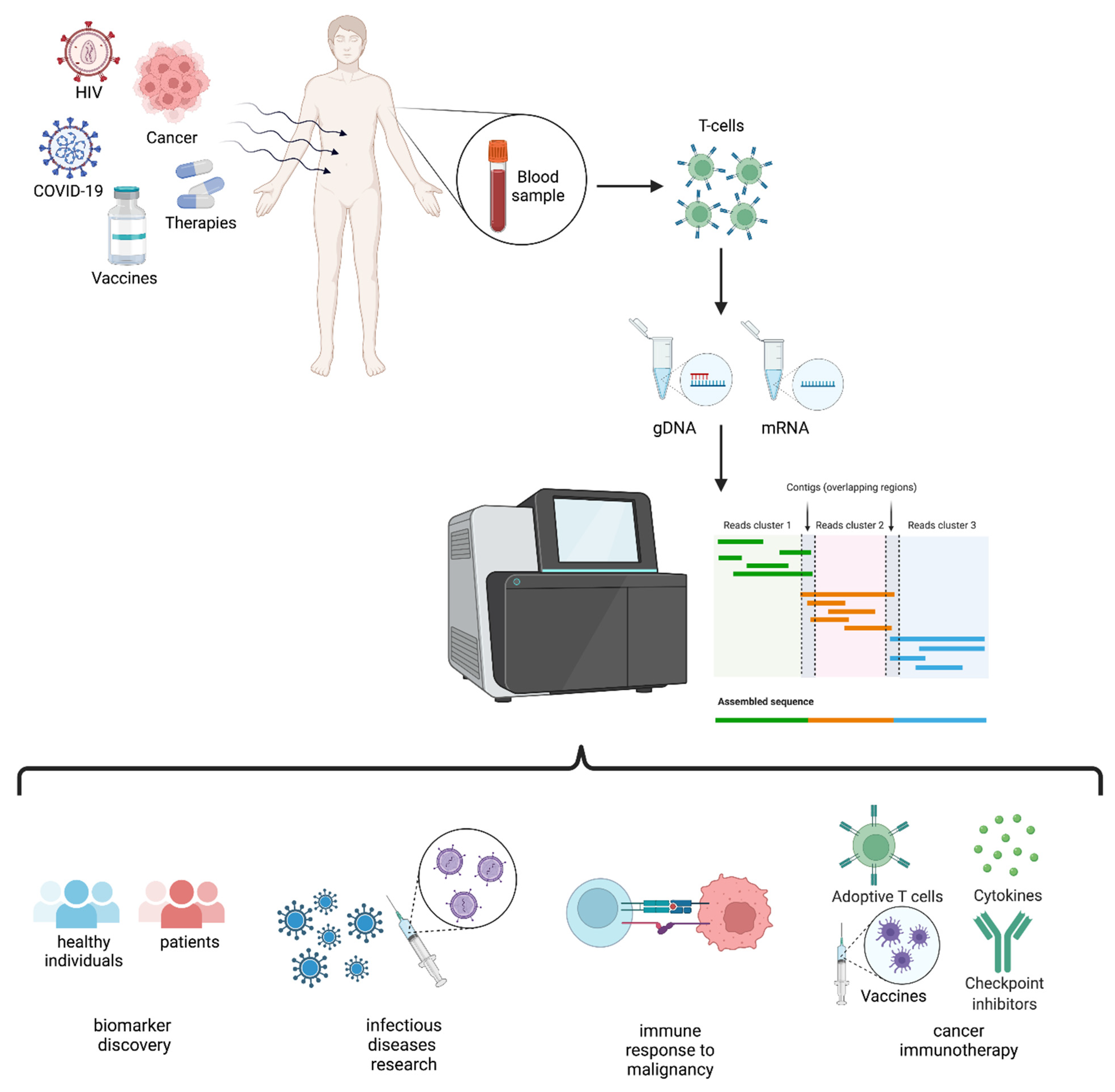
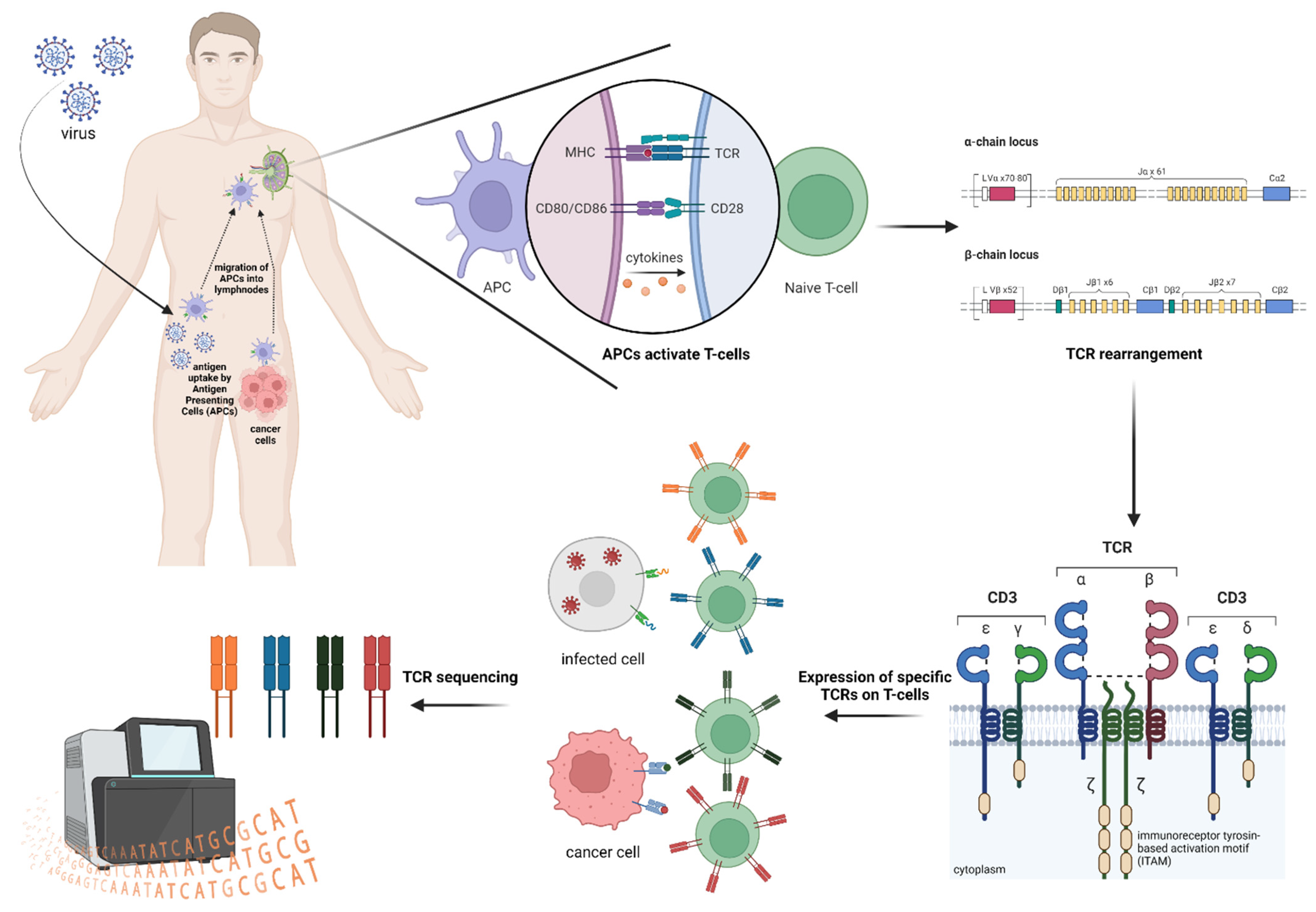
2. TCR Repertoire Analysis
3. TCR and HLA
| Type of HLA Alleles Association | HLA Typing Future Opportunities | Example |
|---|---|---|
| With specific infectious diseases or the severity of infection | To provide insight into differences in T-cell repertoires in infectious disease and patterns of T-cell targeting | Heterozygous individuals progress less rapidly to AIDS than HLA homozygous individuals after HIV infection [31]. Kaslow et al. found that HLA-B27 and B57 were strongly associated with slow progression to AIDS [36]. |
| With increased risk of or protection from various autoimmune disorders | To clarify a subject’s disease state and potentially stratify patients for treatment studies. | Association of the HLA class I region has been detected for several autoimmune diseases (AIDs); some examples are: |
| With cancer therapy outcomes | To understand and infer the efficacy of immunotherapy in specific individuals | Higher heterozygosity in HLA has been linked to a better response to anti-cancer treatments [41]. |
4. TCR Repertoire via HTS: When Details Matter
| Advantages | |
| gDNA | mRNA |
|
|
| Disadvantages | |
| gDNA | mRNA |
|
|
References
- Liu, H.; Pan, W.; Tang, C.; Tang, Y.; Wu, H.; Yoshimura, A.; Deng, Y.; He, N.; Li, S. The Methods and Advances of Adaptive Immune Receptors Repertoire Sequencing. Theranostics 2021, 11, 8945–8963.
- Dahal-Koirala, S.; Balaban, G.; Neumann, R.S.; Scheffer, L.; Lundin, K.E.A.; Greiff, V.; Sollid, L.M.; Qiao, S.-W.; Sandve, G.K. TCRpower: Quantifying the Detection Power of T-Cell Receptor Sequencing with a Novel Computational Pipeline Calibrated by Spike-in Sequences. Brief. Bioinform. 2022, 23, bbab566.
- Li, N.; Yuan, J.; Tian, W.; Meng, L.; Liu, Y. T-cell Receptor Repertoire Analysis for the Diagnosis and Treatment of Solid Tumor: A Methodology and Clinical Applications. Cancer Commun. 2020, 40, 473–483.
- Aversa, I.; Malanga, D.; Fiume, G.; Palmieri, C. Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. Int. J. Mol. Sci. 2020, 21, 2378.
- Pai, J.A.; Satpathy, A.T. High-Throughput and Single-Cell T Cell Receptor Sequencing Technologies. Nat. Methods 2021, 18, 881–892.
- Watkins, T.S.; Miles, J.J. The Human T-cell Receptor Repertoire in Health and Disease and Potential for Omics Integration. Immunol. Cell Biol. 2021, 99, 135–145.
- Chiffelle, J.; Genolet, R.; Perez, M.A.; Coukos, G.; Zoete, V.; Harari, A. T-Cell Repertoire Analysis and Metrics of Diversity and Clonality. Curr. Opin. Biotechnol. 2020, 65, 284–295.
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of Methodologies for T-Cell Receptor Repertoire Analysis. BMC Biotechnol. 2017, 17, 61.
- Yamauchi, M. Mechanisms Underlying the Suppression of Chromosome Rearrangements by Ataxia-Telangiectasia Mutated. Genes 2021, 12, 1232.
- Nishana, M.; Raghavan, S.C. Role of Recombination Activating Genes in the Generation of Antigen Receptor Diversity and Beyond. Immunology 2012, 137, 271–281.
- Yang, G.; Ou, M.; Chen, H.; Guo, C.; Chen, J.; Lin, H.; Tang, D.; Xue, W.; Li, W.; Sui, W.; et al. Characteristic Analysis of TCR β-Chain CDR3 Repertoire for Pre- and Post-Liver Transplantation. Oncotarget 2018, 9, 34506–34519.
- Bassing, C.H.; Swat, W.; Alt, F.W. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 2002, 109, S45–S55.
- Calis, J.J.A.; Rosenberg, B.R. Characterizing Immune Repertoires by High Throughput Sequencing: Strategies and Applications. Trends Immunol. 2014, 35, 581–590.
- Valkiers, S.; de Vrij, N.; Gielis, S.; Verbandt, S.; Ogunjimi, B.; Laukens, K.; Meysman, P. Recent Advances in T-Cell Receptor Repertoire Analysis: Bridging the Gap with Multimodal Single-Cell RNA Sequencing. ImmunoInformatics 2022, 5, 100009.
- Pauken, K.E.; Lagattuta, K.A.; Lu, B.Y.; Lucca, L.E.; Daud, A.I.; Hafler, D.A.; Kluger, H.M.; Raychaudhuri, S.; Sharpe, A.H. TCR-Sequencing in Cancer and Autoimmunity: Barcodes and Beyond. Trends Immunol. 2022, 43, 180–194.
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddel, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive Assessment of T-Cell Receptor β-Chain Diversity in Aβ T Cells. Blood 2009, 114, 4099–4107.
- Warren, R.L.; Freeman, J.D.; Zeng, T.; Choe, G.; Munro, S.; Moore, R.; Webb, J.R.; Holt, R.A. Exhaustive T-Cell Repertoire Sequencing of Human Peripheral Blood Samples Reveals Signatures of Antigen Selection and a Directly Measured Repertoire Size of at Least 1 Million Clonotypes. Genome Res. 2011, 21, 790–797.
- Nikolich-Žugich, J.; Slifka, M.K.; Messaoudi, I. The Many Important Facets of T-Cell Repertoire Diversity. Nat. Rev. Immunol. 2004, 4, 123–132.
- Dupic, T.; Marcou, Q.; Walczak, A.M.; Mora, T. Genesis of the Aβ T-Cell Receptor. PLoS Comput. Biol. 2019, 15, e1006874.
- Sewell, A.K. Why Must T Cells Be Cross-Reactive? Nat. Rev. Immunol. 2012, 12, 669–677.
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213.
- Gutierrez, L.; Beckford, J.; Alachkar, H. Deciphering the TCR Repertoire to Solve the COVID-19 Mystery. Trends Pharmacol. Sci. 2020, 41, 518–530.
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular Regulation of Effector and Memory T Cell Differentiation. Nat. Immunol. 2014, 15, 1104–1115.
- Daniels, M.A.; Teixeiro, E. TCR Signaling in T Cell Memory. Front. Immunol. 2015, 6, 617.
- Zvyagin, I.V.; Pogorelyy, M.V.; Ivanova, M.E.; Komech, E.A.; Shugay, M.; Bolotin, D.A.; Shelenkov, A.A.; Kurnosov, A.A.; Staroverov, D.B.; Chudakov, D.M.; et al. Distinctive Properties of Identical Twins’ TCR Repertoires Revealed by High-Throughput Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 5980–5985.
- Chen, G.; Yang, X.; Ko, A.; Sun, X.; Gao, M.; Zhang, Y.; Shi, A.; Mariuzza, R.A.; Weng, N. Sequence and Structural Analyses Reveal Distinct and Highly Diverse Human CD8 + TCR Repertoires to Immunodominant Viral Antigens. Cell Rep. 2017, 19, 569–583.
- Pantaleo, G.; Demarest, J.F.; Soudeyns, H.; Graziosi, C.; Denis, F.; Adelsberger, J.W.; Borrow, P.; Saag, M.S.; Shaw, G.M.; Sekaly, R.P.; et al. Major Expansion of CD8+ T Cells with a Predominant vp Usage during the Primary Immune Response to HIV. Nature 1994, 370, 463–467.
- Li, B.; Li, T.; Pignon, J.-C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of Tumor-Infiltrating T Cell Repertoire of Human Cancers. Nat. Genet. 2016, 48, 725–732.
- Holtmeier, W.; Kabelitz, D.; Gammadelta, T. Cells Link Innate and Adaptive Immune Responses. Chem. Immunol. Allergy 2005, 86, 151–183.
- Woodsworth, D.J.; Castellarin, M.; Holt, R.A. Sequence Analysis of T-Cell Repertoires in Health and Disease. Genome Med. 2013, 5, 98.
- Mosaad, Y.M. Clinical Role of Human Leukocyte Antigen in Health and Disease. Scand. J. Immunol. 2015, 82, 283–306.
- DeWitt, W.S.; Smith, A.; Schoch, G.; Hansen, J.A.; Matsen, F.A.; Bradley, P. Human T Cell Receptor Occurrence Patterns Encode Immune History, Genetic Background, and Receptor Specificity. eLife 2018, 7, e38358.
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA Variation and Disease. Nat. Rev. Immunol. 2018, 18, 325–339.
- Amiel, J. Study of the Leukocyte Phenotypes in Hodgkin’s Disease in Histocompatibility Testing; Teraski, P.I., Ed.; Munksgaard: Copenhagen, Denmark, 1967; pp. 79–81.
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2013, 14, 301–323.
- Kaslow, R.A.; Carrington, M.; Apple, R.; Park, L.; Muñoz, A.; Saah, A.J.; Goedert, J.J.; Winkler, C.; O’Brien, S.J.; Rinaldo, C.; et al. Influence of Combinations of Human Major Histocompatibility Complex Genes on the Course of HIV-1 Infection. Nat. Med. 1996, 2, 405–411.
- Simmonds, M.; Gough, S. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465.
- Flores-Robles, B.-J.; Labrador-Sánchez, E.; Andrés-Trasahedo, E.; Pinillos-Aransay, V.; Joven-Zapata, M.-Y.; Torrecilla Lerena, L.; Salazar-Asencio, O.-A.; López-Martín, J.-A. Concurrence of Rheumatoid Arthritis and Ankylosing Spondylitis: Analysis of Seven Cases and Literature Review. Case Rep. Rheumatol. 2022, 2022, 8500567.
- Padyukov, L. Genetics of Rheumatoid Arthritis. Semin. Immunopathol. 2022, 44, 47–62.
- Abdul-Hussein, S.S.; Ali, E.N.; Zaki, N.H.; Ad’hiah, A.H. Genetic Polymorphism of HLA-G Gene (G*01:03, G*01:04, and G*01:05N) in Iraqi Patients with Inflammatory Bowel Disease (Ulcerative Colitis and Crohn’s Disease). Egypt J. Med. Hum. Genet. 2021, 22, 34.
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587.
- De Simone, M.; Rossetti, G.; Pagani, M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front. Immunol. 2018, 9, 1638.
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46.
- Rizzo, J.M.; Buck, M.J. Key Principles and Clinical Applications of “Next-Generation” DNA Sequencing. Cancer Prev. Res. 2012, 5, 887–900.
- Takara Bio Blog Team. 4 Factors to Consider for Immune Repertoire Profiling; Web Document Reprint; Takara Bio USA, Inc.: San Jose, CA, USA, 2019.
- Kockelbergh, H.; Evans, S.; Deng, T.; Clyne, E.; Kyriakidou, A.; Economou, A.; Luu Hoang, K.N.; Woodmansey, S.; Foers, A.; Fowler, A.; et al. Utility of Bulk T-Cell Receptor Repertoire Sequencing Analysis in Understanding Immune Responses to COVID-19. Diagnostics 2022, 12, 1222.
- Barennes, P.; Quiniou, V.; Shugay, M.; Egorov, E.S.; Davydov, A.N.; Chudakov, D.M.; Uddin, I.; Ismail, M.; Oakes, T.; Chain, B.; et al. Benchmarking of T Cell Receptor Repertoire Profiling Methods Reveals Large Systematic Biases. Nat. Biotechnol. 2021, 39, 236–245.
- Shugay, M.; Britanova, O.V.; Merzlyak, E.M.; Turchaninova, M.A.; Mamedov, I.Z.; Tuganbaev, T.R.; Bolotin, D.A.; Staroverov, D.B.; Putintseva, E.K.; Plevova, K.; et al. Towards Error-Free Profiling of Immune Repertoires. Nat. Methods 2014, 11, 653–655.
- Logan, A.C.; Gao, H.; Wang, C.; Sahaf, B.; Jones, C.D.; Marshall, E.L.; Buno, I.; Armstrong, R.; Fire, A.Z.; Weinberg, K.I.; et al. High-Throughput VDJ Sequencing for Quantification of Minimal Residual Disease in Chronic Lymphocytic Leukemia and Immune Reconstitution Assessment. Proc. Natl. Acad. Sci. USA 2011, 108, 21194–21199.
- Tiller, T.; Busse, C.E.; Wardemann, H. Cloning and Expression of Murine Ig Genes from Single B Cells. J. Immunol. Methods 2009, 350, 183–193.
- Li, S.; Wilkinson, M.F. Nonsense Surveillance in Lymphocytes? Immunity 1998, 8, 135–141.
- Wang, C.; Liu, Y.; Xu, L.T.; Jackson, K.J.L.; Roskin, K.M.; Pham, T.D.; Laserson, J.; Marshall, E.L.; Seo, K.; Lee, J.; et al. Effects of Aging, Cytomegalovirus Infection, and EBV Infection on Human B Cell Repertoires. J. Immunol. 2014, 192, 603–611.
- Mamedov, I.Z.; Britanova, O.V.; Zvyagin, I.V.; Turchaninova, M.A.; Bolotin, D.A.; Putintseva, E.V.; Lebedev, Y.B.; Chudakov, D.M. Preparing Unbiased T-Cell Receptor and Antibody CDNA Libraries for the Deep Next Generation Sequencing Profiling. Front. Immunol. 2013, 4, 456.
- Trück, J.; Eugster, A.; Barennes, P.; Tipton, C.M.; Luning Prak, E.T.; Bagnara, D.; Soto, C.; Sherkow, J.S.; Payne, A.S.; Lefran, M.; et al. Biological Controls for Standardization and Interpretation of Adaptive Immune Receptor Repertoire Profiling. eLife 2021, 10, e66274.
- Yip, S.H.; Sham, P.C.; Wang, J. Evaluation of Tools for Highly Variable Gene Discovery from Single-Cell RNA-Seq Data. Brief. Bioinform. 2019, 20, 1583–1589.
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57.
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by Shrinking: Empowering Single-Cell “omics” with Microfluidic Devices. Nat. Rev. Genet. 2017, 18, 345–361.
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317.
- Salomon, R.; Kaczorowski, D.; Valdes-Mora, F.; Nordon, R.E.; Neild, A.; Farbehi, N.; Bartonicekcg, N.; Gallego-Ortega, D. Droplet-Based Single Cell RNAseq Tools: A Practical Guide. Lab Chip 2019, 19, 1706–1727.
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2022, 12, 802080.
- Carlson, C.S.; Emerson, R.O.; Sherwood, A.M.; Desmarais, C.; Chung, M.-W.; Parsons, J.M.; Steen, M.S.; LaMadrid-Herrmannsfeldt, M.A.; Williamson, D.W.; Livingston, R.J.; et al. Using Synthetic Templates to Design an Unbiased Multiplex PCR Assay. Nat. Commun. 2013, 4, 2680.
- Wulf, M.G.; Maguire, S.; Humbert, P.; Dai, N.; Bei, Y.; Nichols, N.M.; Correa, I.R., Jr.; Guan, S. Non-Templated Addition and Template Switching by Moloney Murine Leukemia Virus (MMLV)-Based Reverse Transcriptases Co-Occur and Compete with Each Other. J. Biol. Chem. 2019, 294, 18220–18231.
- Adamopoulos, P.G.; Tsiakanikas, P.; Stolidi, I.; Scorilas, A. A Versatile 5′ RACE-Seq Methodology for the Accurate Identification of the 5′ Termini of MRNAs. BMC Genom. 2022, 23, 163.
- Liu, X.; Zhang, W.; Zeng, X.; Zhang, R.; Du, Y.; Hong, X.; Cao, H.; Su, Z.; Wang, C.; Wu, J.; et al. Systematic Comparative Evaluation of Methods for Investigating the TCRβ Repertoire. PLoS ONE 2016, 11, e0152464.
- Aoki, H.; Shichino, S.; Matsushima, K.; Ueha, S. Revealing Clonal Responses of Tumor-Reactive T-Cells Through T Cell Receptor Repertoire Analysis. Front. Immunol. 2022, 13, 807696.
- Ye, J.; Ma, N.; Madden, T.L.; Ostell, J.M. IgBLAST: An Immunoglobulin Variable Domain Sequence Analysis Tool. Nucleic Acids Res. 2013, 41, W34–W40.
- Alamyar, E.; Duroux, P.; Lefranc, M.P.; Giudicelli, V. IMGT ® Tools for the Nucleotide Analysis of Immunoglobulin (IG) and T Cell Receptor (TR) V- (D)-J Repertoires, Polymorphisms, and IG Mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 2012, 882, 569–604.
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat. Methods 2015, 12, 380–381.
- Weber, C.R.; Akbar, R.; Yermanos, A.; Pavlović, M.; Snapkov, I.; Sandve, G.K.; Reddy, S.T.; Greiff, V. ImmuneSIM: Tunable Multi-Feature Simulation of B- and T-Cell Receptor Repertoires for Immunoinformatics Benchmarking. Bioinformatics 2020, 36, 3594–3596.
- Gerritsen, B.; Pandit, A.; Andeweg, A.C.; de Boer, R.J. RTCR: A Pipeline for Complete and Accurate Recovery of T Cell Repertoires from High Throughput Sequencing Data. Bioinformatics 2016, 32, 3098–3106.


