 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesco Feo | + 2996 word(s) | 2996 | 2020-10-10 03:59:00 | | | |
| 2 | Catherine Yang | Meta information modification | 2996 | 2020-10-21 05:49:47 | | |
Video Upload Options
The pioneering observation by Otto Warburg that an elevated glucose consumption by cancer cells is associated with a restraint of oxygen consumption and elevated aerobic glycolysis, induced several researches on the molecular changes involved in the metabolic deregulation of cancer cells. This review analyzes the relationships between the deregulationn of respiration and glycolysis, the defective cancer mitochondria, the molecular and biochemical alterations involved in cancer pathogenesis and progression and new approaches to cancer therapy aimed at the correction of the molecular and metabolic changes characterizing cancer cells.
1. Introduction
Following the pioneering observation by Otto Warburg that an elevated glucose consumption by cancer cells is associated with a restraint of oxygen consumption and the production of lactic acid in aerobiosis (Warburg effect) [1][2], numerous efforts were dedicated to explain the genesis of the glycolytic metabolism of tumors as well as its implication in the malignant transformation. The analysis of a spectrum of hepatocellular carcinoma (HCC), including well, moderately, and poorly differentiated tumors, showed that aerobic glycolysis increases with the degree of malignancy [3][4], suggesting that the Warburg effect is correlated to tumor progression.
The role of Warburg effect in the pathogenesis of tumors has been the object of debate in recent years [3][4]. However, recent acquisitions on cancer biochemistry and biology contributed to clarify the pathogenesis of the Warburg effect and its role in tumor progression, and suggested new approaches to HCC therapy.
2. The Warburg Effect and Tumor Therapy
According to available data, cancer is one of the deadliest health problems. New therapeutic treatments should address the major factors implied in the resistance of tumors to standard treatments. Metabolic deviations can vary among different types of cancers and different patients. The tumor heterogeneity requires strategies targeting the various metabolic deviations of cancers. Nowadays, metabolic-oriented research offers different new perspectives, which may contribute to the development of innovative and effective therapeutic treatments.
2.1. Therapeutic Effect of the Glycolysis Inhibition
Previous work in our laboratory showed a decrease of SAM liver content during the development of preneoplastic nodules and HCC induced in rats by diethylnitrosamine. The treatment of rats with SAM reconstituted the SAM pool and inhibited the growth of nodules and HCCs [5]. We further observed [6] that the administration of SAM causes a consistent fall of the number and DNA synthesis of preneoplastic liver lesions associated to a decrease in liver PK, LDH and GPDH (Glycerol-3-phosphate dehydrogenase). SAM did not affect these enzymatic activities in normal liver, but caused a consistent decrease in initiated rats. Enolase, FBP, and ME activities also increased in the liver of initiated rats, but were not significantly affected by SAM. These results clearly showed the association of the inhibition of some glycolytic enzymes with the arrest of the growth of preneoplastic and neoplastic liver lesions. They were in accordance with the previous demonstration that the inhibitor of HK and GPI, by 2-DG [7], completely hampers the aerobic glycolysis and protein synthesis of the hepatoma ascites AH-130 but is ineffective in the rat bone marrow cells and cells isolated from chicken embryo, where the energy necessary for the protein synthesis is given by respiration. As expected, 2-DG inhibits the glycolysis and protein synthesis of all tissues tested in anaerobiosis [7]. In accordance with these findings, the inhibition of HK2 in human liver tumor Huh7 and HepG2 cells inhibits cell growth and increases cell death [8]. Furthermore, 2-DG induces apoptosis of neuroblastoma SK-N-BE (2) cells [9] and its chronic dietary administration inhibits tumor incidence in a mouse model of hepatocarcinogenesis [10]. Moreover, 2-DG synergizes with Newcastle disease virus to kill breast cancer cells to inhibit GA-3-P dehydrogenase [11], and increases the efficacy of Adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo [12].
Enzo et al. observed that active glycolysis is needed for the full activity of the transcriptional cofactor YAP and TAZ (Yes-associated protein/transcriptional coactivator with PDZ-binding motif) and the primary human mammary tumors with active YAP/TAZ progress toward more advanced malignant stages [13]. YAP and TAZ are two transcriptional coactivators that promote cell proliferation through a transcriptional program intermediated by TEAD transcription factor [14]. It was also found that YAP1 is genetically controlled in rat liver cancer and determines the fate and stem-like behavior of the human disease [15]. YAP increases glycolysis [12] and supports the expression of Glut3, a known driver of a cancer stem cell phenotype, whose expression is elevated in cancer [15][16]. YAP/TAZ/TEAD and AP-1 form a complex that synergistically activates target genes controlling S-phase entry and mitosis [17]. In addition, in HepG2, Huh7, and Hep3B cells, forced YAP1 over-expression results in the expression of stem cell markers and increases cell viability [15]. This does not occur if YAP1 expression is inhibited by specific siRNA or the cells are transfected with mutant YAP1 that does not bind to TEAD [15].
PFK catalyzes the rate-limiting phosphorylation of F6P to F-1,6-BP, an energy consuming step of glycolysis (Figure 1). One small molecule, PFK15, inhibitor of PFAKFB3, suppresses glucose uptake and growth in Lewis lung carcinomas in syngeneic mice and has anti-tumor effects in three human xenograft models of cancer in athymic mice [18]. Furthermore, the inhibition of PFKFB3 impedes glucose uptake, glycolysis, and growth of HER2-expressing breast cancer cells [19]. Treatment with lapatinib, an FDA-approved HER2 inhibitor, decreases PFKFB3 expression and glucose metabolism in HER2+ cells [19] In vivo administration of a PFKFB3 antagonist significantly suppresses the growth of HER2-driven breast tumors [19]. The competitive inhibitors of FKFB3, 3PO and PFK15, decrease glucose metabolism and the proliferation of different cancer cells and inhibit the growth, metastatic spread and glucose metabolism in three human xenograft models of cancer in athymic mice [20]. Finally, also GAPDH is a potential cancer therapeutic target as well, since inhibition of this glycolytic enzyme by 2-DG inhibits cancer cell growth [21].
Attempts by Shikonin to inhibit of PKM2 have shown a significant inhibition of glycolysis in tumors [22]. However, this compound is highly toxic and poorly soluble. Better results were obtained with metformin, less toxic, which increases the sensitivity to chemotherapeutic drugs by inhibiting PKM2 [23].
The inhibition of LDH is another proposed strategy to arrest tumor growth. Specifically it has been shown that LDH suppression induces oxidative stress and reduces tumor progression [24][25]. Its association to the inhibition of four glycolysis pathway molecules (GLUT1, HKII, PFKFB3, PDHK1) using WZB117, 3PO, 3-bromopyruvate, Dichloroacetate inhibitors (Phloretin, Quercetin, STF31Oxamic acid, NHI-1) results in an increase of extracellular glucose, a decrease of lactate production and a rise in apoptosis of cancer cells [26]. In MIA PaCa-2 human pancreatic cells, the inhibition of LDHA by GNE-140 for 2 days was found to trigger cell death, although the activation of the AMPK-mTOR-S6K signaling induced resistance to GNE-140 and increased oxidative phosphorylation [27]. Thus, the combination of the LDHA inhibitor with compounds targeting the AMPK-mTOR-S6K signaling was proposed as a potential strategy to reduce the emergence of resistance to LDHA inhibition [28]. Also, oxamate significantly suppresses the proliferation of NSCLC cells, while it exerts a much lower toxicity in normal cells [29]. Moreover, the use of miR-30a-5p, an LDHA inhibitor, has been proposed as another possible strategy to counteract the resistance to chemical inhibitors [24].
The inhibition of the lactic acid transporters MCT1/2 slows down cancer cell proliferation [30][31][32]. Mechanistic studies of MCT1 inhibitors suggest that loss of LDHA activity may generate ROS through negative feedback effects on glycolysis and the synthesis of GSH [33]. Cancer cells do not metabolize lactate but export it thus inducing the acidification of the tumor surrounding [34]. The induction of inflammation by lactate efflux attracts immune cells including macrophages. The secretion of macrophage cytokines and growth factors if not reach deadly concentrations for cancer cells, may stimulate tumor cell growth and metastasis formation [35][36]. It should be noted that MCT (monocarboxylate transporter) also functions for lactate import into cells that oxidize lactate, such as heart and skeletal muscle, or metabolize for gluconeogenesis such as liver and kidney [37]. Low MCT levels are present in many issues, but marked increases in the MCT1 and/or MCT4 levels occur in several human tumors, which thus might be treated with MCTs inhibitors.
Clinical trials with glycolytic inhibitors have also been performed [38] with 2-DG (trials I/II), the HK inhibitors Lonidamine (trials II/III) and 3-Bromopyruvate (pre-clinical), Imatinib, an inhibitor of Bcr-Abl tyrosine kinase that causes a decrease in HK and G6PD activities (approved for clinical use), and Oxythiamine, which suppresses pentose phosphate pathway by inhibiting transketolase and inhibits pyruvate dehydrogenase. The above findings clear indicate the contribution of glycolytic energy to the growth of cancer cells thus confirming the old findings [39] on the therapeutic effect of the glycolytic inhibitor 2-DG.
In a recent interesting review, Cassim and coworkers [40] report that mitochondria play a key function in tumorigenesis. To explain the role of LDH in tumor growth, these authors disrupted the LDHA and LDHB genes in the human colon adenocarcinoma and murine melanoma cells [41]. The knockout of each of these genes did not strongly reduced lactate secretion, which was instead fully suppressed in the double knockout cells (LDHA/B-DKO). Under normoxia, LDHA/B-DKO cells survived by shifting their metabolism to oxidative phosphorylation (OXPHOS), leading to a 2-fold decrease in proliferation rates both in vitro and in vivo. However, under hypoxia the LDHA/B suppression completely abolished in vitro growth, indicating its dependency on OXPHOS. These conditions were reproduced pharmacologically by treating WT cells with the LDHA/B-specific inhibitor GNE-140. These findings demonstrate that the Warburg effect is not only based on the high expression of LDHA, since both LDHA and LDHB need to be deleted to suppress fermentative glycolysis. Furthermore, on the basis of these findings, the Warburg effect does not seem to be indispensable even in aggressive tumors. Hence, according to the authors, the mitochondrial metabolism should be a target of innovative anti-cancer agents. However, the problem of the translation of preclinical drugs, effective in eradicating cancer cells, to the clinical use still remains. Indeed, these drugs would necessarily affect normal cells, including anti-cancer immune cells. Therefore, new therapeutic strategies devoted to the modulation of mitochondrial functions in a distinct cellular target need to be defined.
2.2. Intracellular Alkalinity of Cancer Cells
Recently, the Warburg effect has been explained as an effect of the intracellular alkalinity of cancer cells [42]. It was found that the glycolytic activation is itself an oncogenic event: indeed, the overexpression of the glucose transporter GLUT3 in nonmalignant human breast cells suffices to activate oncogenic signaling pathways such as EGFR, β1 integrin, MEK, and AKT, with consequent loss of tissue polarity and increased growth [42]. A proton [H+]-related mechanism involved in the initiation and progression of cancer has been recently described [43][44]. Specifically, it was shown that, in tumor cells, an extracellular acid microenvironment of 6.2–6.9 vs 7.3–7.4 in normal cells is associated with alkaline intracellular pH values of 7.12–7.7 vs 6.99–7.05 in normal cells [45][46][47]. This situation is linked to an elevated activity of the membrane-bound Na+/H+ exchanger isoform 1 (NHE1) [48][49]. Therefore, the use of NHE1 inhibitors with low side-effects such as Cariporide and other more potent NHE1 inhibitors, including compound 9t, have been recently proposed for cancer therapy [49].
2.3. Glutaminolysis and Cancer Therapy
Glutamine has some important functions in tumor metabolism and cancer cells depend on glutamine metabolism. Therefore, targeting glutamine metabolism could contribute to cancer therapy. Allosteric inhibitors of GLS (glutaminase) have shown promising results in cancer models. One of these inhibitors, BAPTES [bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide] [50][51], inhibits the proliferation of cancer cells in vitro, and of xenografts in vivo, and prolongs survival in genetically engineered mouse models of cancer [52][53]. In addition, BAPTES increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice [54]. Preclinical research has shown that CB-839, a GLS inhibitor, is efficacious against breast cancer and hematological malignancies [55][56].
GLS inhibition has been also found to be efficient in combination therapy. The inhibition of the anti-apoptotic protein BCL-2 integrates glutaminase inhibition [57]. Furthermore, it has been shown that highly invasive ovarian cancer cells have increased glutamine dependence compared to less invasive cells [58], and metastatic prostate tumors show increased dependence on glutamine uptake [58]. Furthermore, increased lactate in tumor microenvironment may promote increased glutamine metabolism though HIF2 and MYC-dependent mechanism [59], and increased ammonia released from cancer cells stimulates autophagy in the fibroblasts that consequently release additional glutamine, which is metabolized by the cancer cells [60] but may be toxic to surrounding cells. Interestingly, the lactate produced in cancer cells by increased glycolysis, which may be present in the tumor microenvironment, promotes glutamine metabolism by a HIF2 and MYC-dependent mechanism [61][62]. Increased glutaminolysis produces ammonia with consequent autophagy of exposed cells [63][64], such as fibroblasts, and further release of glutamine, which may be metabolized by cancer cells [65]. It must be also noted that some tumors, including those of brain and lung [66][67][68][69] synthesize and excrete glutamine. Ammonia may be toxic in this context and is probably detoxified by surrounding non-transformed cells.
Glutaminolysis inhibition could be curative for highly invasive ovarian cancer cells exhibiting increased glutamine dependence compared to less invasive cancers [70]. Similarly, metastatic prostate tumors show increased dependence on glutamine uptake [68][69][71]. Furthermore, the genetic inhibition of glutaminase was shown to prevent epithelial-to-mesenchymal transition, a key step in tumor cell invasiveness and eventual metastasizing [72].
2.4. Nampt Inhibitors
Nampt has been considered a potential therapeutic target for cancer treatment, due to its contribution to cancer pathogenesis [73]. Nampt converts NAM (nicotinamide) and PRPP (5-phosphoribose-1-pyrophosphate) to nicotinamide mononucleotide, a substrate of Nmnat (nicotinamide mononucleotide adenylyl transferase) to generate NAD (nicotinamide adenine dinucleotide) by transferring the adenylyl moiety (Figure 2). An alternative way to generate NAD is the transformation of nicotinic acid (NA) to NAMN (nicotinic acid mononucleotide), catalyzed by Naprt (nicotinic acid phosphoribosyltransferase), followed by the synthesis of NAAD (nicotinic acid adenine dinucleotide), in presence of ATP, catalyzed by Nmnat (nicotinamide mononucleotide adenylyltransferase) and the transformation of NAAD to NA, catalyzed by a synthetase (Figure 2). Different Nampt inhibitors have been developed to date, among them FK866 (APO866) [74] reduces cellular levels of NAD and GAPDH [75][76], inhibits glycolysis and cancer cell growth [77] and induces apoptosis [74]. Another competitive Nampt inhibitor, GMX1778, has anti-cancer effects [78]. A recent study demonstrated that recurrent NAMPT-H191R mutations confer resistance to treatment of cancer cell lines with TF-31, but STF-31 also exerts an additional inhibitory effect against Nampt [79]. Although Nampt inhibition may prevent tumor cell growth, it is essential for normal cells and its absence may cause embryonic lethality, progressive muscle degeneration [80][81] and severe vision loss [82]. It must be noted, however, that Naprt (nicotinate phosphoribosyltransferase) generates nicotinic acid mononucleotide (NAMN) from nicotinic acid and PRPP (5-phosphoribose-1-pyrophosphate), and then Nmnat (nicotinamide mononucleotide adenylyltransferase) conjugates ATP to NAMN to generate NAAD (nicotinic acid adenine dinucleotide) (Figure 2) [83]. IDH (isocitrate dehydrogenase) mutations in glioma cell lines induce a decrease of Naprt expression via an increase of DNA histone methylations [84]. IDH mutations are frequent in patients with gliomas and acute myeloid leukemia (AML) [85]. Mutated IDH aberrantly generates 2-hydroxyglutarate that inhibits DNA and histone demethylases and promotes the hypermethylation of DNA histones [85].
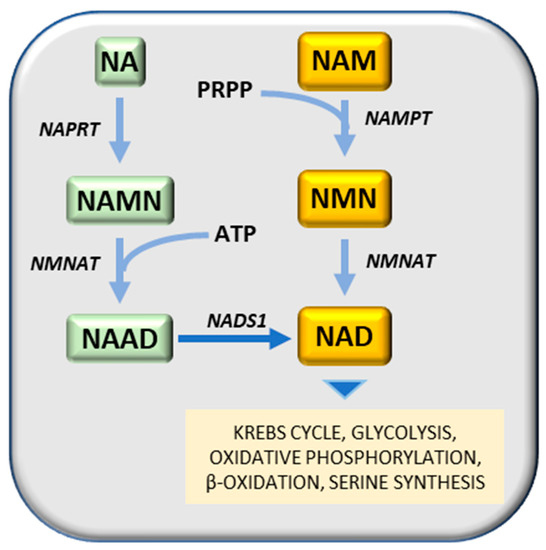
Figure 2. NAD synthesis. Substrates: NA, nicotinic acid; NAD, Nicotinamide adenine dinucleotide; NAAD, nicotinic acid adenine dinucleotide; NAM, nicotinamide; NAMN, nicotinic acid mononucleotide; NMN, nicotinamide mononucleotide; PRFPP, 5-phosphoribose-1-pyrophosphate. Enzymes: NADS1, NAD synthetase; NAMPT, nicotinamide phosphoribosyltransferase; NMNAT, nicotinamide mononucleotide adenylyltransferase; NAPRT, nicotinic acid phosphoribosyltransferase.
3. Conclusions
In 1861, Louis Pasteur observed that oxygen increased the division of yeast and the inhibition of the fermentation by oxygen. In 1913 the mitochondria were discovered, and Otto Warburg linked cellular respiration to these organelles, derived from guinea pig liver extracts, that he called “grana” [86]. Warburg observed that tumors acquire the unusual property of fermenting glucose to lactate in the presence of oxygen (aerobic glycolysis), and proposed that deficient mitochondrial respiration causes aerobic glycolysis in cancer cells [86]. However, we now know that tumor mitochondria are well coupled (Figure 2) [87][88][89] and the Pasteur defect is largely linked to the great increase in the activity of glycolytic enzymes in cancer cells.
The fermentation of glucose to lactate in the presence of oxygen (Warburg effect) has been strongly criticized by Weinhouse [90] who, in opposition to the Warburg effect, observed that the well differentiated Morris hepatomas do not produce lactic acid in aerobiosis. Indeed, an almost complete absence of lactic acid production in four well-differentiated tumors has been found, and in aerobiosis, relatively low production in three well differentiated tumors, and high production in poorly-differentiated tumors (Figure 3). Altogether, these observations indicate that the Warburg effect is not the cause of the malignant transformation but its consequence, and an adaptation to hypoxia of tumor cells that becomes more evident during cancer progression (see the paragraph 3).
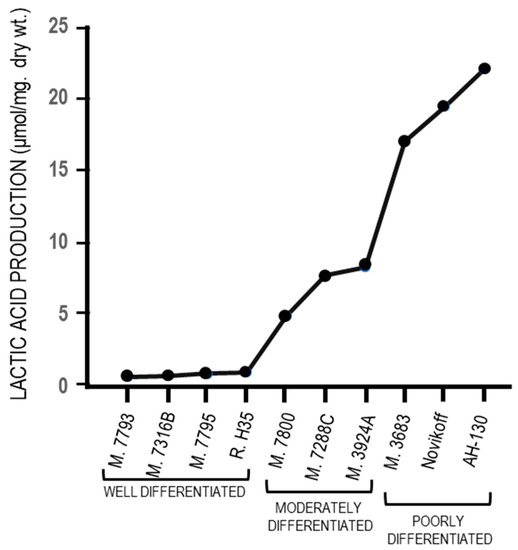
Figure 3. Aerobic glycolysis in hepatocellular carcinomas with different levels of differentiation.
The first report on the Warburg effect was published 97 years ago [1][2]. Thirty-four years later the primary metabolic block produced by 2-DG [91] was localized, and forty years after the Warburg report of 1923, the inhibition of aerobic glycolysis by 2-DG [39] was demonstrated, as an inhibitor of HK and GPI [91], which inhibited protein synthesis in cancer cells. Some important recent studies reevaluate the old hypotheses. A series of elegant experiments has unequivocally shown that glycolytic metabolism activates the YAP/TAZ signal in cells of different neoplasms (e.g., breast and liver) and YAP activation upregulates the expression and transcriptional activity of HK2 and PFKFB3 [38][40]. Through the use of glycolysis inhibitors, it was observed that the YAP/TAZ signal, active in cells that incorporate glucose and produce lactic acid, is strongly inhibited when glucose metabolism is blocked, or glycolysis is reduced. It has been shown that the glycolytic enzyme PFK1, upregulated in tumors, promotes the YAP/TAZ transcriptional cooperation with TEAD factors to form the PFK1-TEAD1-YAP/TAZ complex in the nucleus. YAP activation during glycolysis, mediated by the hexosamine biosynthesis pathway, and its acetylation by O-linked b-N-acetylglucosamine transferase promote its nuclear translocation and transcriptional activity [15][92][93]. YAP/TEAD in the cell nucleus regulates GLUT3 transcription and activates YAP, thus augmenting HK2 expression and promoting glycolysis [15][93]. Therefore, the glycolytic metabolism regulates the transcription of YAP/TAZ [94] that promotes glycolysis, lipogenesis, and glutaminolysis, and is involved in the proliferative activity and aggressivity of neoplastic cells [15][93]. Collectively, this body of evidence clearly indicates that the Warburg effect is an epiphenomenon of the transformation process essential for the development of malignancy.
References
- Warburg, O. Versuche an überlebendem carcinom-gewebe. Klin. Wochenschr. 1923, 2, 776–777.
- Warburg, O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137.
- Elwood, J.; Lin, Y.; Cristofalo, V.; Weinhouse, S.; Morris, H.P. Glucose utilization in homogenates of the Morris hepatoma 5123 and related tumors. Cancer Res. 1963, 23, 906–913.
- Weinhouse, S. Glycolysis, respiration, and anomalous gene expression in experimental hepatomas. G.H.A. Clowes memorial lecture. Cancer Res. 1972, 32, 2007–2016.
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622.
- Pascale, R.M.; Marras, V.; Simile, M.M.; Daino, L.; Pinna, G.; Bennati, S.; Carta, M.; A Seddaiu, M.; Massarelli, G.; Feo, F. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: A long-term study. Cancer Res. 1992, 52, 4979–4986.
- Gerbracht, U.; Eigenbrodt, E.; Simile, M.M.; Pascale, R.M.; Gaspa, L.; Daino, L.; A Seddaiu, M.; De Miglio, M.R.; Nufris, A.; Feo, F. Effect of S-adenosyl-L-methionine on the development of preneoplastic foci and the activity of some carbohydrate metabolizing enzymes in the liver, during experimental hepatocarcinogenesis. Anticancer. Res. 1993, 13, 1965–1972.
- Terranova, T.; Feo, F.; Gravela, E.; Gariel, L. Die wirkung der 2-desoxyglucose auf energhetischen stoffwechsel nund auf die proteinsynthese von tumorzellen und normalzellen. Z. Krebsforsch 1964, 66, 41–45.
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.-M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446.
- Bertero, T.; Cottrill, K.A.; Annis, S.; Bhat, B.; Gochuico, B.R.; Osorio, J.C.; Rosas, I.; Haley, K.J.; Corey, K.E.; Chung, R.T. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci. Rep. 2015, 5, 18277.
- Singh, S.; Pandey, S.; Bhatt, A.N.; Chaudhary, R.; Bhuria, V.; Kalra, N.; Soni, R.; Roy, B.G.; Saluja, D.; Dwarakanath, B. Chronic Dietary Administration of the Glycolytic Inhibitor 2-Deoxy-D-Glucose (2-DG) Inhibits the Growth of Implanted Ehrlich’s Ascites Tumor in Mice. PLoS ONE 2015, 10, e0132089.
- Al-Shammari, A.M.; Abdullah, A.H.; Allami, Z.M.; Yaseen, N.Y. 2-Deoxyglucose and Newcastle Disease Virus Synergize to Kill Breast Cancer Cells by Inhibition of Glycolysis Pathway Through Glyceraldehyde3-Phosphate Downregulation. Front. Mol. Biosci. 2019, 6, 90.
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34.
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP / TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370.
- Koo, J.H.; Guan, K.-L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206.
- Naito, A.; Cook, C.C.; Mizumachi, T.; Wang, M.; Xie, C.H.; Evans, T.T.; Kelly, T.; Higuchi, M. Progressive tumor features accompany epithelial-mesenchymal transition induced in mitochondrial DNA-depleted cells. Cancer Sci. 2008, 99, 584–1588.
- Simile, M.M.; Latte, G.; Demartis, M.I.; Brozzetti, S.; Calvisi, D.F.; Porcu, A.; Feo, C.F.; Seddaiu, M.A.; Daino, L.; Berasain, C.; et al. Post-translational deregulation of YAP1 is genetically controlled in rat liver cancer and determines the fate and stem-like behavior of the human disease. Oncotarget 2016, 7, 49194–49216.
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227.
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A.; Klarer, A.C.; Redman, R.; Miller, N.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470.
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40.
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120.
- Krasnov, G.S.; Dmitriev, A.A.; Snezhkina, A.V.; Kudryavtseva, A.V. Deregulation of glycolysis in cancer: Glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert Opin. Ther. Targets 2013, 17, 681–693.
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int. J. Boil. Sci. 2018, 14, 1883–1891.
- Lei, R.; Zhang, S.; Wang, Y.; Dai, S.; Sun, J.; Zhu, C. Metformin Inhibits Epithelial-to-Mesenchymal Transition of Keloid Fibroblasts via the HIF-1α/PKM2 Signaling Pathway. Int. J. Med Sci. 2019, 16, 960–966.
- Granchi, C.; Bertini, S.; Macchia, M.; Minutolo, F. Inhibitors of lactate dehydrogenase isoforms and their therapeutic potentials. Curr. Med. Chem. 2010, 17, 672–697.
- Oshima, N.; Ishida, R.; Kishimoto, S.; Beebe, K.; Brender, J.R.; Yamamoto, K.; Urban, D.; Rai, G.; Johnson, M.S.; Benavides, G.; et al. Dynamic Imaging of LDH Inhibition in Tumors Reveals Rapid In Vivo Metabolic Rewiring and Vulnerability to Combination Therapy. Cell Rep. 2020, 30, 1798.e4–1810.e4.
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695.
- Boudreau, A.; E Purkey, H.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Methods 2016, 12, 779–786.
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget 2014, 5, 11886–11896.
- Li, L.; Kang, L.; Zhao, W.; Feng, Y.; Liu, W.; Wang, T.; Mai, H.; Huang, J.; Chen, S.; Liang, Y.; et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017, 400, 89–98.
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.-P.; Pouysségur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668.
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920.
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta Bioenerg. 2016, 1866, 151–162.
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1323.
- Wike-Hooley, J.L.; Haveman, J.; Reinhold, H.S. The relevance of tumour pH to the treatment of malignant disease. Radiother. Oncol. 1984, 2, 343–366.
- Yabu, M.; Shime, H.; Hara, H.; Saito, H.; Matsumoto, M.; Seya, T.; Akazawa, T.; Inoue, N. IL-23-dependent and -independent enhancement pathways of IL-17A production by lactic acid. Int. Immunol. 2011, 23, 29–41.
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183.
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family—Role and regulation. IUBMB Life 2012, 64, 109–119.
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637.
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006, 25, 4633–4646.
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119.
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Boil. Chem. 2018, 293, 15947–15961.
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.B.; Elhassan, G.O.; Ibrahim, M.E.; Orozco, J.D.P.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802.
- Reshkin, S.J.; Cardone, R.A.; Harguindey, S. Na+-H+ exchanger, pH regulation and cancer. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 85–99.
- Brisson, L.; Reshkin, S.J.; Goré, J.; Roger, S. pH regulators in invadosomal functioning: Proton delivery for matrix tasting. Eur. J. Cell Boil. 2012, 91, 847–860.
- Brisson, L.; Gillet, L.; Calaghan, S.C.; Besson, P.; Le Guennec, J.-Y.; Roger, S.; Gore, J. NaV1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H+ efflux in caveolae. Oncogene 2010, 30, 2070–2076.
- Harguindey, S.; Arranz, J.L.; Wahl, M.L.; Orive, G.; Reshkin, S.J. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer. Res. 2009, 29, 2127–2136.
- Doppler, W.; Jaggi, R.; Groner, B. Induction of v-mos and activated Ha-ras oncogene expression in quiescent NIH 3T3 cells causes intracellular alkalinisation and cell-cycle progression. Gene 1987, 54, 147–153.
- Wang, H.; Long, X.; Wang, D.; Lou, M.; Zou, D.; Chen, R.; Nian, W.; Zhou, Q. Increased expression of Na+/H+ exchanger isoform 1 predicts tumor aggressiveness and unfavorable prognosis in epithelial ovarian cancer. Oncol Lett. 2018, 16, 6713–6720.
- Atwal, K.S.; O’Neil, S.V.; Ahmad, S.; Doweyko, L.; Kirby, M.; Dorso, C.R.; Chandrasena, G.; Chen, B.-C.; Zhao, R.; Zahler, R. Synthesis and biological activity of 5-aryl-4-(4-(5-methyl-1H-imidazol-4-yl)piperidin-1-yl)pyrimidine analogs as potent, highly selective, and orally bioavailable NHE-1 inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 4796–4799.
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414.
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121.
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306.
- Colombo, S.L.; Palacios-Callender, M.; Frakich, N.; Carcamo, S.; Kovacs, I.; Tudzarova, S.; Moncada, S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc. Natl. Acad. Sci, USA 2011, 108, 21069–21074.
- Boysen, G.; Jamshidi-Parsian, A.; Davis, M.A.; Siegel, E.R.; Simecka, C.M.; Kore, R.A.; Dings, R.P.M.; Griffin, R.J. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Boil. 2019, 95, 436–442.
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901.
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2018, 62, 1096–1115.
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728.
- Wang, Q.; Hardie, R.-A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289.
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, C.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201.
- Mariño, G.; Kroemer, G. Ammonia: A Diffusible Factor Released by Proliferating Cells That Induces Autophagy. Sci. Signal. 2010, 3, pe19.
- Perez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83.
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126.
- Eng, C.H.; Yu, K.; Lucas, J.; White, E.; Abraham, R.T. Ammonia Derived from Glutaminolysis Is a Diffusible Regulator of Autophagy. Sci. Signal. 2010, 3, ra31.
- Ko, Y.-H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Boil. Ther. 2011, 12, 1085–1097.
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.P.; Higashi, R.M.; Miller, N.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698.
- Choi, C.; Ganji, S.; Hulsey, K.; Madan, A.; Kovacs, Z.; Dimitrov, I.; Zhang, S.; Pichumani, K.; Mendelsohn, D.; Mickey, B.; et al. A comparative study of short- and long-TE ¹H MRS at 3 T for in vivo detection of 2-hydroxyglutarate in brain tumors. NMR Biomed. 2013, 26, 1242–1250.
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.I.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837.
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694.
- Jacque, N.; Ronchett, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Trovati, M.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356.
- Dasgupta, S.; Putluri, N.; Long, W.; Zhang, B.; Wang, J.; Kaushik, A.; Arnold, J.M.; Bhowmik, S.K.; Stashi, E.; Brennan, C.A.; et al. Coactivator SRC-2-dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J. Clin. Investig. 2015, 125, 1174–1188.
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939.
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17.
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442.
- Tan, B.; Young, D.A.; Lu, Z.-H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.-S.; et al. Pharmacological Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, in Human Cancer Cells. J. Boil. Chem. 2012, 288, 3500–3511.
- Tan, B.; Dong, S.; Shepard, R.L.; Kays, L.; Roth, K.D.; Geeganage, S.; Kuo, M.-S.; Zhao, G. Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, Leads to Altered Carbohydrate Metabolism in Cancer Cells. J. Boil. Chem. 2015, 290, 15812–15824.
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the Central Metabolite NAD Leads to Oncosis-mediated Cell Death. J. Boil. Chem. 2014, 289, 35182–35192.
- Gehrke, I.; Bouchard, E.D.J.; Beiggi, S.; Poeppl, A.G.; Johnston, J.B.; Gibson, S.; Banerji, V. On-Target Effect of FK866, a Nicotinamide Phosphoribosyl Transferase Inhibitor, by Apoptosis-Mediated Death in Chronic Lymphocytic Leukemia Cells. Clin. Cancer Res. 2014, 20, 4861–4872.
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT Is the Cellular Target of STF-31-Like Small-Molecule Probes. ACS Chem. Boil. 2014, 9, 2247–2254.
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375.
- Frederick, D.W.; Loro, E.; Liu, L.; Davila, A., Jr.; Chellappa, K.; Silverman, I.M.; Frederick, D.W.; Loro, E.; Liu, L.; Davila, A., Jr.; et al. Loss of NAD homeostasis leads to progressive and reversibledegeneration of skeletal muscle. Cell Metab. 2016, 24, 269–282.
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Séne, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016, 17, 69–85.
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of Cellular NAD Levels by Nicotinic Acid and Involvement of Nicotinic Acid Phosphoribosyltransferase in Human Cells. J. Boil. Chem. 2007, 282, 24574–24582.
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784.
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, Ş.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Chance, B.; Garfinkel, D.; Higgins, J.; Hess, B. Metabolic control mechanisms. A solution for the equations representing interaction between glycolysis and respiration in ascites tumor cells. J. Biol. Chem. 1960, 235, 2426–2439.
- Frau, M.; Simile, M.M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.M.E.; Seddaiu, M.A.; Brozzetti, S.; Feo, C.F.; Massarelli, G.; Solinas, G.; et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell Oncol. 2012, 35, 163–173.
- Jiang, X.; Sun, Q.; Li, H.; Li, K.; Ren, X. The role of phosphoglycerate mutase 1 in tumor aerobic glycolysis and its potential therapeutic implications. Int. J. Cancer 2013, 135, 1991–1996.
- Weinhouse, S. Hepatomas. Science 1967, 158, 542–543.
- Wick, A.N.; Drury, D.R.; I Nakada, H.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Boil. Chem. 1957, 224, 963–969.
- Zheng, X.; Han, H.; Liu, G.; Ma, Y.; Pan, R.; Sang, L.; Li, R.; Yang, L.; Marks, J.R.; Wang, W.; et al. Lnc RNA wires up Hippo and Hedgehog signaling to reprogramme glucose metabolism. EMBO J. 2017, 36, 3325–3335.
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The role of YAP/TAZ activity in cancer metabolic reprogramming. Mol. Cancer 2018, 17, 134.
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.K.; Lin, Z.; Balliet, R.M.; Howell, A.; et al. Understanding the “lethal” drivers of tumor stroma co-evolution: Emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Boil. Ther. 2010, 10, 537–542.


