Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrice Forget | -- | 2104 | 2022-08-18 03:34:17 | | | |
| 2 | Catherine Yang | Meta information modification | 2104 | 2022-08-18 03:37:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mokini, Z.; Cama, A.; Forget, P. Anesthetics and Long Term Cancer Outcomes. Encyclopedia. Available online: https://encyclopedia.pub/entry/26259 (accessed on 05 July 2026).
Mokini Z, Cama A, Forget P. Anesthetics and Long Term Cancer Outcomes. Encyclopedia. Available at: https://encyclopedia.pub/entry/26259. Accessed July 05, 2026.
Mokini, Zhirajr, Alessandro Cama, Patrice Forget. "Anesthetics and Long Term Cancer Outcomes" Encyclopedia, https://encyclopedia.pub/entry/26259 (accessed July 05, 2026).
Mokini, Z., Cama, A., & Forget, P. (2022, August 18). Anesthetics and Long Term Cancer Outcomes. In Encyclopedia. https://encyclopedia.pub/entry/26259
Mokini, Zhirajr, et al. "Anesthetics and Long Term Cancer Outcomes." Encyclopedia. Web. 18 August, 2022.
Copy Citation
Anesthetics are chemical factors with the potential to induce epigenetic effects. With regard to the nervous system, for example, this is supported by the fact that anesthetics are neurotoxins sharing many molecular mechanisms of action with alcohol and cocaine.
cancer
anesthesia
epigenetics
ketorolac
lidocaine
1. Epigenetics
The biological response of cells under the influence of physiological and external factors is driven by epigenetic modulation of gene and protein expression. To make an analogy, genes are the constitution and epigenetic changes are all genetic mechanisms other than DNA sequence changes. Epigenetics are laws, regulatory acts and amendments, or the heritable cellular system that interprets the genome [1][2][3][4][5].
In normal cells, the transcriptional status of most genes, the coding part of the genome, is epigenetically fixed through the binding of DNA with histones. However, other genes reside in a balanced state sensitive to exogenous signals and capable of rapid modulation of DNA transcription to mRNA. These events lead to alterations in gene expression, a consequent differential production of messenger RNAs (mRNAs) and a subsequent modified expression of proteins that are the final biologic effectors. Several short- or long-time-operating factors including external chemical or pharmacological stimuli can alter this balance [6][7]. One of the most significant examples of the effect of epigenetics is synap
1.1. Epigenetics and Anesthetics
Research on the epigenetic effects of anesthetics has been mainly focused on the neurodevelopmental brain alterations and ischemia reperfusion injury effects. The first microarray investigation on inhalational anesthetics showed that expression of 1.5% of 10,000 genes in various organs was altered [8]. Sevoflurane has the ability to change the expression of the circadian genes and drug metabolizing enzymes [9][10]. Propofol and sevoflurane show a different protein expression change activity in rat brains [11]. The effects are more pronounced at young ages, when the central nervous system and other tissues are highly susceptible to what is called epigenetic reprogramming [12][13][14][15]. Anesthetics are also capable of inducing prolonged and intergenerational epigenetic effects. One week after bariatric surgery under general anesthesia, DNA methylation of 1509 genes in male spermatozoa remains altered and 1004 of those genes remained altered after 1 year [16]. Reduced DNA methylation in the 5-upstream promoter region of rat mothers exposed to 6 h sevoflurane and upregulation of Arc and JunB mRNA expression, two genes regulating synaptic plasticity and neuronal development, are trans-generationally expressed in offspring male born [17].
1.2. Post-Translational Chromatin Regulation
Post-translational epigenetic modifications on histones and chromatin are regulated by more than 700 enzymes categorized as writers/erasers, readers, movers shapers and insulators. Lysine-rich N-terminal histone “tails” undergo several processes such as acetylation and methylation, ubiquitination, phosphorylation, and sumoylation [18].
Histone acetylation by acetyltransferases allows chromatin liberation and gene transcription, while histone deacetylation by deacetylases results in a stronger histone bond with DNA and inhibition of gene transcription [19]. Repeated sevoflurane exposure of neonatal rats leads to an increased hippocampal deacetylase activity and reduced histone acetylation, with developmental effects that are reversed by histone deacetylase inhibitors [20].
Methylation by DNA methyltransferases (DNMTs) can both induce and repress gene transcription, depending on the chromatin residue modified. Sevoflurane neurotoxicity on rat stem cells is exerted through a concentration-dependent DNA methylation [21]. There is also an increasingly growing evidence of the epigenetic effects of anesthetics on the developing human brain that include both methylation and acetylation [22].
DNA methylation is one of the mechanisms affected by stress and involved in postoperative hyperalgesia [23]. DNA methylation and DNMT expression in skin after incision is changed and controls nociceptive sensitization. DNMT inhibition attenuates incision-induced nociceptive hypersensitivity via up-regulation of Oprm1 gene expression while treatment with naloxone exacerbates incision-induced mechanical hypersensitivity [23].
DNA methylation in blood mononuclear cells after general anesthesia for major breast surgery is globally reduced by 26% while DNMT mRNAs expression is reduced by 65 to 71% [24]. Opiates are thought to increase methylation whereas lidocaine show controversial effects [25][26][27]. These findings support the evidence that anesthesia/surgery may alter the epigenetic status of host’s tissues after surgery.
Aberrant DNA methylation leads to onco-suppressor gene silencing or oncogene activation and has been linked to oncogenesis in a number of tumor types. Cancers present hypermethylation of CpG islands (DNA regions with a high density of cytosine–guanine dinucleotides) in or near promoter regions, whereas gene bodies are hypomethylated.
1.3. RNA
Only 1.5% of mammalian DNA encodes for approximately 20,000 genes which will be translated into proteins. Protein synthesis is driven by three main types of RNA: messenger RNA (mRNA), transfer RNA (tRNA) that accounts for the majority of RNA molecules, and ribosomal RNA (rRNA). mRNA is transcribed directly from DNA and contains the “message” that is translated by tRNAs into proteins. rRNA accounts for 90% of total RNA mass and forms ribosomes, which are the protein synthesizers [28]. The rest of the human genome, more than 90%, represents non-coding RNAs (ncRNA). The total number of RNA molecules is estimated at 107 per cell [29].
Non-coding RNAs are classified into long [>200 nucleotides] and short ncRNAs (<200 nucleotides) (lncRNA and sncRNA respectively). A series of biological functions such as regulation of the expression of coding genes, cancero-genesis and regulation of biological processes have been described for many ncRNAs, supporting the hypothesis that they are functional, although for most of them a biological role has not yet been discovered [30][31].
Micro RNAs (miRNA) are non-coding, endogenous and highly conserved sncRNAs across species. They act via post-transcriptional degradation or translational repression by binding to 3′ untranslated regions (UTR) of target mRNAs. Each molecule of mRNA can be regulated by more the one miRNA, and a single miRNA may influence the expression of a wide range of mRNAs [32]. Some miRNAs are expressed ubiquitously, whereas others are tissue-specific and/or stage-specific. miRNAs regulate the activity of 30–50% of protein-coding genes and modulate the expression of 10–30% of human genome [33][34]. RNAs also exist extracellularly in the circulation in exosomes [34].
Anesthetics affect miRNA in various cells across the body. In mice, sevoflurane and propofol affect expression of 46 of 177 miRNAs in liver, 20 miRNAs in lung, and 14 miRNAs in brain, with a specific pattern of expression at each anesthetic exposure [35][36][37]. Sevoflurane inhibits the NF-κB pathway through miRNA-9-5p expression and protects the liver from ischemia-reperfusion injury [38]. Sevoflurane also induces miRNA-155 downregulation that reduces systemic inflammation in acute lung injury models [39]. Propofol attenuates the neuroinflammation induced by lipopolysaccharide through miRNA-155 suppression [40].
Given these demonstrated effects on epigenetics, is it biologically plausible that anesthetics affect cancer’s natural history [41]? It is well known that epigenetic modulation in normal tissues may contrast or favor tumor progression, and anesthesia has the ability to change the epigenetics of surgical tissues [24][25][26][27][42]. Can anesthesia also directly alter cancer’s epigenetics? The question as to which mechanism can drive the anesthetic-induced alterations of tumor biology and host systems during a limited period of time such as the perioperative period, to the point that tumor behavior and its future clinical history change permanently, may have a response in epigenetics. However, to date there is no clear response a to whether it is the extent of surgery or stress response rather than anesthesia management that influences most cancer outcomes.
2. Anesthesia and Cancer Epigenetics
Epigenetics has a crucial role in cancero-genesis, which is a complex biological event. An altered gene and protein expression profile may affect cancer cells and the host response, inducing more or less favourable conditions for tumor progression. Cancer growth itself is driven by a “Darwinian” evolutionary process that starts in normal tissues where advantageous genetic and epigenetic events occur in a series of stages that promote clonal expansions of new tissue. Epigenetic changes can be subject to the same selection forces as genetic events [43]. This “new” tissue progressively misses the features of the original tissue. Every aspect of tumor biology including cell growth and differentiation, cell cycle control, cancer stem cell formation, DNA repair, angiogenesis, migration, and evasion of host immune surveillance is affected by epigenetic alterations [44][45].
Only few sporadic studies have investigated the role of anesthesia on cancer cell epigenetics and much is yet to be discovered (Figure 1). Differential gene expression was shown after ex-vivo exposure of brain cancer cell line SH-SY5Y and of breast cancer cell line MCF-7 to enflurane, isoflurane, desflurane, halothane, sevoflurane, and nitrous oxide [46].
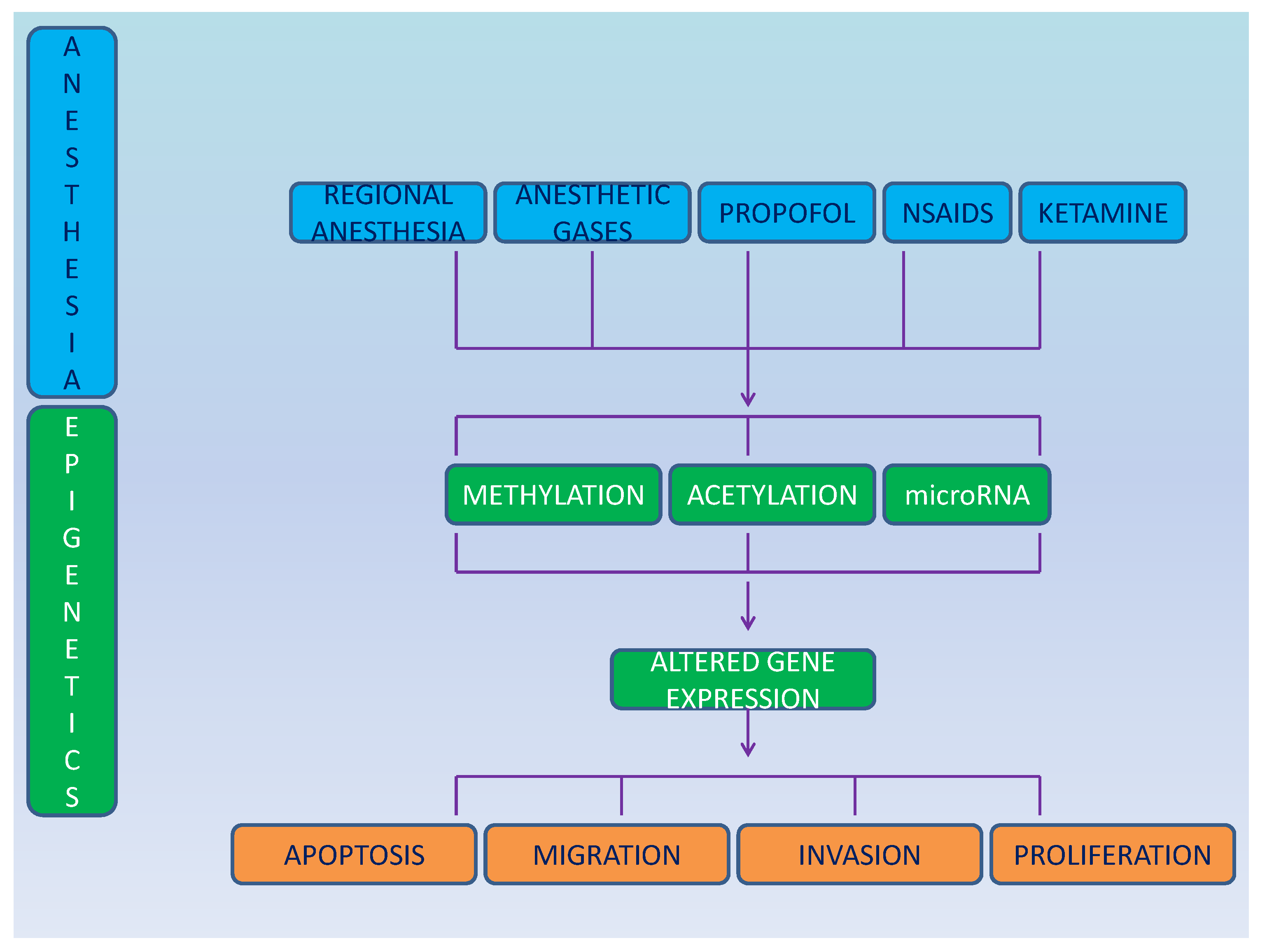
Figure 1. Schematic representation of the effect of anesthesia on epigenetics and cancer cells.
Sevoflurane suppresses viability, invasion, migration, and apoptosis of colorectal cancer cells in a dose-dependent fashion by regulating the circ-HMGCS1/miRNA-34a-5p/SGPP1 axis, via inactivation of the Ras/Raf/MEK/ERK signaling, via regulation of ERK/MMP-9 pathway by up-regulating miRNA-203 and by regulating miRNA-34a/ADAM10 axis [47][48][49][50]. A study also showed a differential and specific impact on circulating exo-somial miRNAs during colon cancer surgery resection [51].
Other anesthetics such as the non-steroidal anti-inflammatory drugs, propofol and ketamine, may also modulate epigenetics. Celecoxib inhibits osteosarcoma cell proliferation, migration, and invasion via miRNA-34a [52]. miRNA-126-5p, −320a and -146a-5p regulate the sensitivity to celecoxib [53].
R-Ketorolac inhibits the activity of Rac1 and Cdc42, which are GTP-ases involved in cell growth and cell cycle regulation, in ovarian cancer patients. Both are believed to be therapeutic targets for ovarian cancer as regulators of migration, adhesion and invasion [54][55]. Studying epigenetics after ketorolac use may help in understanding the mechanism underlying this effect.
Lidocaine at clinical concentrations (1 mM) induced DNA demethylation for 120 h on BT-20 and MCF-7 breast cancer cells in vitro [56]. Lidocaine also reduces the proliferation of pancreatic cancer PaTu8988t cells after 48 h in vitro [57].
Ketamine, a N-methyl-Daspartate [NMDA] receptor antagonist used as a racemic derivative of two enantiomers s[+]ketamine and r[−]ketamine, can also modulate epigenetics. Exposure for 48 h with ketamine and s-ketamine of PaTu8988t pancreatic carcinoma cells significantly inhibited proliferation and expression of nuclear NFATc2 [57]. PaTu8988t and Panc-1 cells express the NMDA type R2a receptor. Ketamine and s-ketamine at 1000 μM concentration for 48 h significantly inhibited the proliferation of pancreatic cancer cells. S-ketamine reduced apoptosis after 3 h in PaTu8988t cells and in PANC-1 cells after 24 h incubation with ketamine [a] (65 ± 17%) and s-ketamine [b] (68 ± 24%). Necrosis increased after 16 h with ketamine and after 6–24 h after s-ketamine [58]. However, clinical doses of ketamine (2 mg/kg iv) produce an average plasma concentration of 41 μg/kg/min, which corresponds to a plasma concentration of 9.3 μM [58]. Ketamine has been recently found to suppress the viability of liver cancer cells and induce ferroptosis through the lncPVT1/miRNA-214-3p/GPX4 pathway [59]. In ovarian cancer, ketamine modulates the P300-mediated H3K27 acetylation activation in the promoter of lncRNA PVT1 that binds histone methyltransferase enhancer of zeste homolog 2 [EZH2], and regulates the expression of target genes, including p57, consequently regulating ovarian cancer cell growth, cell cycle control, apoptosis and colony forming [60].
Propofol (2,6-diisopropylphenol) is an extensively-used sedative anesthetic at 4-20 mcg/mL brain concentrations. Propofol regulates both miRNAs and lncRNAs, and modulates the signaling pathways of important oncogenes/onco-suppressors that are potential therapeutic targets including hypoxia-inducible factor-1α (HIF-1α), mitogen-activated protein kinase (MAPK), nuclear factor-kappaB (NF-κB), and nuclear factor E2-related factor-2 (Nrf2) [61][62].
Propofol upregulates miRNA-34a expression and induces miRNA-34a-dependent apoptosis in SH-SY5Y neuroblastoma cells in vitro [63][64]. At a concentration of 20 μg/mL for 72 h, propofol significantly reduces cell viability and apoptosis in human pancreatic PANC-1 cells increasing the expression of pro-apoptotic caspase-3 and Bax and down-regulating the expression of anti-apoptotic Bcl-2 gene. Apoptosis is induced by miRNA-3-4a-dependent upregulation of LOC285194 [65]. Additionally, propofol inhibits cell growth and metastasis by enhancing miR-328 expression in pancreatic cancer [66]. Propofol also induces a miRNA-34a-dependent E-cadherin upregulation in the PANC-1 cells, and reduces motility of cells is wound healing assays [65].
In human pancreatic Miapaca-2 and Panc-1 cells in vitro and murine pancreatic cancer cell (Panc02) in vivo, propofol shows a concentration-dependent (5, 25, 50, 100 μM) inhibition of cell migration, expression of VEGF and HIF-1α, phosphorylation of (ERK), AKT, (CaMK II), and Ca2+ concentration [67]. Propofol downregulates VEGF and suppresses migration of pancreatic cancer cells by inhibiting the NMDA receptor [67].
Propofol at concentrations of 1 to 10 μg/mL for 48 and 72 h also suppresses the proliferation and invasion of PANC-1 cells by 2.46- and 3.95-fold by upregulating miRNA-133a expression [68][69]. Apoptosis also increases after 1–10 microg/mL propofol exposure in a dose and time-dependent manner and through PUMA pathway [68][69].
Propofol inhibits the growth, invasion and migration of PANC-1 cells in a dose and time-dependent manner via the miRNA-21/Slug pathway and through E-cadherin upregulation [69]. Both PUMA and E-cadherin are upregulated by propofol via miRNA-21 inactivation and subsequent Slug inhibition [69].
The activation and expression of ADAM8 in response to hypoxia in PC is inhibited, thus antagonizing angiogenesis [70]. The downregulation of ADAM8 and upregulation of miRNA-328 mediated by propofol was shown to inhibit pancreatic cancer proliferation and metastasis [71]. Growth of xenograft pancreatic cancer is also inhibited in nude mice models by propofol [70]. Propofol shows a synergistic effect with gemcitabine through downregulation of NF-κB signaling pathway induced by gemcitabine, thereby promoting the chemosensitivity of PC [72].
References
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080.
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705.
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254.
- Ozanne, E.S.; Constancia, M. Mechanisms of Disease: The developmental origins of disease and the role of the epigenotype. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 539–546.
- Kangaspeska, S.; Stride, B.; Métivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115.
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50.
- Sakamoto, A.; Imai, J.-I.; Nishikawa, A.; Honma, R.; Ito, E.; Yanagisawa, Y.; Kawamura, M.; Ogawa, R.; Watanabe, S. Influence of inhalation anesthesia assessed by comprehensive gene expression profiling. Gene 2005, 356, 39–48.
- Kobayashi, K.; Takemori, K.; Sakamoto, A. Circadian gene expression is suppressed during sevoflurane anesthesia and the suppression persists after awakening. Brain Res. 2007, 1185, 1–7.
- Nakazato, K.; Yoshida, Y.; Takemori, K.; Kobayashi, K.; Sakamoto, A. Expressions of genes encoding drug-metabolizing enzymes are altered after sevoflurane, isoflurane, propofol or dexmedetomidine anesthesia. Biomed. Res. 2009, 30, 17–24.
- Tsuboko, Y.; Sakamoto, A. Propofol anaesthesia alters the cerebral proteome differently from sevoflurane anaesthesia. Biomed. Res. 2011, 32, 55–65.
- Heindel, J.J.; Balbus, J.; Birnbaum, L.; Brune-Drisse, M.N.; Grandjean, P.; Gray, K.; Landrigan, P.J.; Sly, P.D.; Suk, W.A.; Cory Slechta, D.; et al. Developmental Origins of Health and Disease: Integrating Environmental Influences. Endocrinology 2015, 156, 3416–3421.
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417.
- Gluckman, P.D.; Hanson, A.M.; Mitchell, M.D. Developmental origins of health and disease: Reducing the burden of chronic disease in the next generation. Genome Med. 2010, 2, 14.
- Baird, J.; Jacob, C.; Barker, M.; Fall, C.H.D.; Hanson, M.; Harvey, N.C.; Inskip, H.M.; Kumaran, K.; Cooper, C. Developmental Origins of Health and Disease: A Lifecourse Approach to the Prevention of Non-Communicable Diseases. Healthcare 2017, 5, 14.
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016, 23, 369–378.
- Chastain-Potts, S.E.; Tesic, V.; Tat, Q.L.; Cabrera, O.H.; Quillinan, N.; Jevtovic-Todorovic, V. Sevoflurane Exposure Results in Sex-Specific Transgenerational Upregulation of Target IEGs in the Subiculum. Mol. Neurobiol. 2020, 57, 11–22.
- Bates, S.E. Epigenetic Therapies for Cancer. New Engl. J. Med. 2020, 383, 650–663.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Jia, M.; Liu, W.-X.; Yang, J.-J.; Xu, N.; Xie, Z.-M.; Ju, L.-S.; Ji, M.-H.; Martynyuk, A.E.; Yang, J.-J. Role of histone acetylation in long-term neurobehavioral effects of neonatal Exposure to sevoflurane in rats. Neurobiol. Dis. 2016, 91, 209–220.
- Wang, K.; Tian, Y.; Zhang, Y.; Li, X.; Wei, X.; Hu, H.; Xu, S. Toxicity mechanism of sevoflurane in neural stem cells of rats through DNA methylation. Exp. Ther. Med. 2019, 18, 237–241.
- Cabrera, O.H.; Useinovic, N.; Jevtovic-Todorovic, V. Neonatal anesthesia and dysregulation of the epigenome. Biol. Reprod. 2021, 105, 720–734.
- Sun, Y.; Sahbaie, P.; Liang, D.; Li, W.; Shi, X.; Kingery, P.; Clark, J.D. DNA Methylation Modulates Nociceptive Sensitization after Incision. PLoS ONE 2015, 10, e0142046.
- Caputi, F.F.; Carboni, L.; Rullo, L.; Alessandrini, I.; Balzani, E.; Melotti, R.M.; Romualdi, P.; Candeletti, S.; Fanelli, A. An Exploratory Pilot Study of Changes in Global DNA Methylation in Patients Undergoing Major Breast Surgery under Opioid-Based General Anesthesia. Front. Pharmacol. 2021, 12, 733577.
- Doehring, A.; Oertel, B.G.; Sittl, R.; Lötsch, J. Chronic opioid use is associated with increased DNA methylation correlating with increased clinical pain. Pain 2013, 154, 15–23.
- Fragou, D.; Zanos, P.; Kouidou, S.; Njau, S.; Kitchen, I.; Bailey, A.; Kovatsi, L. Effect of chronic heroin and cocaine administration on global DNA methylation in brain and liver. Toxicol. Lett. 2013, 218, 260–265.
- Lirk, P.; Fiegl, H.; Weber, N.C.; Hollmann, M.W. Epigenetics in the perioperative period. Br. J. Pharmacol. 2015, 172, 2748–2755.
- Liu, Y.; Ding, M.; Gao, Q.; He, A.; Liu, Y.; Mei, H. Current Advances on the Important Roles of Enhancer RNAs in Gene Regulation and Cancer. BioMed Res. Int. 2018, 2018, 2405351.
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2.
- Amin, N.; McGrath, A.; Chen, Y.-P.P. Author Correction: Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2020, 2, 236.
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta. Gene Regul. Mech. 2020, 1863, 194417.
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402.
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J. Cell. Mol. Med. 2019, 23, 7933–7945.
- Ishikawa, M.; Tanaka, S.; Arai, M.; Genda, Y.; Sakamoto, A. Differences in microRNA Changes of Healthy Rat Liver between Sevoflurane and Propofol Anesthesia. Anesthesiology 2012, 117, 1245–1252.
- Tanaka, S.; Ishikawa, M.; Arai, M.; Genda, Y.; Sakamoto, A. Changes in microRNA expression in rat lungs caused by sevoflurane anesthesia: A TaqMan® low-density array study. Biomed. Res. 2012, 33, 255–263.
- Goto, G.; Hori, Y.; Ishikawa, M.; Tanaka, S.; Sakamoto, A. Changes in the gene expression levels of microRNAs in the rat hippocampus by sevoflurane and propofol anesthesia. Mol. Med. Rep. 2014, 9, 1715–1722.
- Liao, X.; Zhou, S.; Zong, J.; Wang, Z. Sevoflurane exerts protective effects on liver ischemia/reperfusion injury by regulating NFKB3 expression via miR-9-5p. Exp. Ther. Med. 2019, 17, 2632–2640.
- Otsuki, T.; Ishikawa, M.; Hori, Y.; Goto, G.; Sakamoto, A. Volatile anesthetic sevoflurane ameliorates endotoxin-induced acute lung injury via microRNA modulation in rats. Biomed. Rep. 2015, 3, 408–412.
- Lu, Y.; Jian, M.-Y.; Ouyang, Y.-B.; Han, R.-Q. Changes in Rat Brain MicroRNA Expression Profiles Following Sevoflurane and Propofol Anesthesia. Chin. Med J. 2015, 128, 1510–1515.
- Martynyuk, E.A.; Ju, L.-S.; Morey, E.T.; Zhang, J.-Q. Neuroendocrine, epigenetic, and intergenerational effects of general anesthetics. World J. Psychiatry 2020, 10, 81–94.
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673.
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28.
- Lund, A.H.; van Lohuizen, M. Epigenetics and cancer. Genes Dev. 2004, 18, 2315–2335.
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386.
- Huitink, J.M.; Heimerikxs, M.; Nieuwland, M.; Loer, S.A.; Brugman, W.; Velds, A.; Sie, D.; Kerkhoven, R.M. Volatile Anesthetics Modulate Gene Expression in Breast and Brain Tumor Cells. Anesth. Analg. 2010, 111, 1411–1415.
- He, J.; Zhao, H.; Liu, X.; Wang, D.; Wang, Y.; Ai, Y.; Yang, J. Sevoflurane suppresses cell viability and invasion and promotes cell apoptosis in colon cancer by modulating exosome-mediated circ-HMGCS1 via the miR-34a-5p/SGPP1 axis. Oncol. Rep. 2020, 44, 2429–2442.
- Yang, X.; Zheng, Y.-T.; Rong, W. Sevoflurane induces apoptosis and inhibits the growth and motility of colon cancer in vitro and in vivo via inactivating Ras/Raf/MEK/ERK signaling. Life Sci. 2019, 239, 116916.
- Fan, L.; Wu, Y.; Wang, J.; He, J.; Han, X. Sevoflurane inhibits the migration and invasion of colorectal cancer cells through regulating ERK/MMP-9 pathway by up-regulating miR-203. Eur. J. Pharmacol. 2019, 850, 43–52.
- Sun, S.Q.; Ren, L.J.; Liu, J.; Wang, P.; Shan, S.M. Sevoflurane inhibits migration and invasion of colorectal cancer cells by regulating microRNA-34a/ADAM10 axis. Neoplasma 2019, 66, 887–895.
- Buschmann, D.; Brandes, F.; Lindemann, A.; Maerte, M.; Ganschow, P.; Chouker, A.; Schelling, G.; Pfaffl, M.W.; Reithmair, M. Propofol and Sevoflurane Differentially Impact MicroRNAs in Circulating Extracellular Vesicles during Colorectal Cancer Resection: A Pilot Study. Anesthesiology 2020, 132, 107–120.
- Chen, X.; Peng, D.; Shen, Y.; Liu, B.; Zhou, H.; Tao, H.; Huang, J. The potential combinational effect of miR-34a with celecoxib in osteosarcoma. Anti-Cancer Drugs 2017, 28, 888–897.
- Dong, Z.; Jiang, H.; Jian, X.; Zhang, W. Change of miRNA expression profiles in patients with knee osteoarthritis before and after celecoxib treatment. J. Clin. Lab. Anal. 2019, 33, e22648.
- Guo, Y.; Kenney, S.R.; Cook, L.; Adams, S.F.; Rutledge, T.; Romero, E.; Oprea, T.I.; Sklar, L.A.; Bedrick, E.; Wiggins, C.L.; et al. A Novel Pharmacologic Activity of Ketorolac for Therapeutic Benefit in Ovarian Cancer Patients. Clin. Cancer Res. 2015, 21, 5064–5072.
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac Targets Cdc42 and Rac1 and Alters Ovarian Cancer Cell Behaviors Critical for Invasion and Metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227.
- Lirk, P.; Berger, R.; Hollmann, M.W.; Fiegl, H. Lidocaine time- and dose-dependently demethylates deoxyribonucleic acid in breast cancer cell lines in vitro. Br. J. Anaesth. 2012, 109, 200–207.
- Malsy, M.; Graf, B.; Bundscherer, A. The Effects of Analgesics and Local Anesthetics on Gene Transcription Mediated by NFATc2 and Sp1 in Pancreatic Carcinoma. Anticancer Res. 2019, 39, 4721–4728.
- Malsy, M.; Gebhardt, K.; Gruber, M.; Wiese, C.; Graf, B.; Bundscherer, A. Effects of ketamine, s-ketamine, and MK 801 on proliferation, apoptosis, and necrosis in pancreatic cancer cells. BMC Anesthesiol. 2015, 15, 111.
- He, G.-N.; Bao, N.-R.; Wang, S.; Xi, M.; Zhang, T.-H.; Chen, F.-S. Ketamine Induces Ferroptosis of Liver Cancer Cells by Targeting lncRNA PVT1/miR-214-3p/GPX4. Drug Des. Dev. Ther. 2021, 15, 3965–3978.
- Li, T.; Yang, J.; Yang, B.; Zhao, G.; Lin, H.; Liu, Q.; Wang, L.; Wan, Y.; Jiang, H. Ketamine Inhibits Ovarian Cancer Cell Growth by Regulating the lncRNA-PVT1/EZH2/p57 Axis. Front. Genet. 2021, 11, 597467.
- Jiang, S.; Liu, Y.; Huang, L.; Zhang, F.; Kang, R. Effects of propofol on cancer development and chemotherapy: Potential mechanisms. Eur. J. Pharmacol. 2018, 831, 46–51.
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Ranjbar, A.; Saleh, S.H.; Bagherian, M.; Sharifzadeh, S.O.; Hushmandi, K.; et al. Regulation of Nuclear Factor-KappaB (NF-κB) signaling pathway by non-coding RNAs in cancer: Inhibiting or promoting carcinogenesis? Cancer Lett. 2021, 509, 63–80.
- Xing, N.; Xing, F.; Li, Y.; Li, P.; Zhang, J.; Wang, D.; Zhang, W.; Yang, J. Dexmedetomidine improves propofol-induced neuronal injury in rat hippocampus with the involvement of miR-34a and the PI3K/Akt signaling pathway. Life Sci. 2020, 247, 117359.
- Li, G.F.; Li, Z.B.; Zhuang, S.J. Inhibition of microRNA-34a protects against propofol anesthesia-induced neurotoxicity and cognitive dysfunction via the MAPK/ERK signaling pathway. Neurosci. Lett. 2018, 675, 152–159.
- Wang, H.; Jiao, H.; Jiang, Z.; Chen, R. Propofol inhibits migration and induces apoptosis of pancreatic cancer PANC-1 cells through miR-34a-mediated E-cadherin and LOC285194 signals. Bioengineered 2020, 11, 510–521.
- Yu, X.; Gao, Y.; Zhang, F. Propofol inhibits pancreatic cancer proliferation and metastasis by up-regulating miR-328 and down-regulating ADAM8. Basic Clin. Pharmacol. Toxicol. 2019, 125, 271–278.
- Chen, X.; Wu, Q.; You, L.; Chen, S.; Zhu, M.; Miao, C. Propofol attenuates pancreatic cancer malignant potential via inhibition of NMDA receptor. Eur. J. Pharmacol. 2017, 795, 150–159.
- Wang, Z.; Gong, H.; Zheng, F.; Liu, D.; Dong, T. Propofol suppresses proliferation and invasion of pancreatic cancer cells by upregulating microRNA-133a expression. Genet. Mol. Res. 2015, 14, 7529–7537.
- Liu, Z.; Zhang, J.; Hong, G.; Quan, J.; Zhang, L.; Yu, M. Propofol Inhibits Growth and Invasion of Pancreatic Cancer Cells through Regulation of the MiR-21/Slug Signaling Pathway. Am. J. Transl. Res. 2016, 8, 4120–4133.
- Gao, Y.; Yu, X.; Zhang, F.; Dai, J. Propofol inhibits pancreatic cancer progress under hypoxia via ADAM 8. J. Hepato-Biliary-Pancreatic Sci. 2019, 26, 219–226.
- Yu, X.; Shi, J.; Wang, X.; Zhang, F. Propofol affects the growth and metastasis of pancreatic cancer via ADAM8. Pharmacol. Rep. 2019, 72, 418–426.
- Du, Q.-H. Propofol induces apoptosis and increases gemcitabine sensitivity in pancreatic cancer cells in vitro by inhibition of nuclear factor-κB activity. World J. Gastroenterol. 2013, 19, 5485–5492.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
555
Revisions:
2 times
(View History)
Update Date:
18 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No