 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Helen Ismail | + 2240 word(s) | 2240 | 2020-10-07 10:39:29 | | | |
| 2 | Vivi Li | -4 word(s) | 2236 | 2020-10-21 06:00:31 | | |
Video Upload Options
Traumatic brain injury (TBI) is a major health concern worldwide and is classified based on severity into mild, moderate, and severe. The mechanical injury in TBI leads to a metabolic and ionic imbalance, which eventually leads to excessive production of reactive oxygen species (ROS) and a state of oxidative stress. To date, no drug has been approved by the food and drug administration (FDA) for the treatment of TBI. Nevertheless, it is thought that targeting the pathology mechanisms would alleviate the consequences of TBI. For that purpose, antioxidants have been considered as treatment options in TBI and were shown to have a neuroprotective effect. Of these, edaravone and mitoquinone seem to be promising.
1. Traumatic Brain Injury: Definition and Pathogenesis
Traumatic brain injury (TBI) is considered a major global health concern with no current FDA-approved drug for its treatment. TBI, defined as “an alteration of brain function, or an evidence of brain pathology, that is caused by an external force” has a major impact on the military personnel and the civilian population as well [1]. The incidence of TBI is estimated to be 939 in 100,000 worldwide with the major causes being falls, vehicle accidents, wars, and sports [2][3][4][5]. The mortality rate of TBI worldwide is estimated to be between 7% and 23% with 90% of TBI-related deaths occurring in developing countries [6][7]. Additionally, TBI imposes an economic burden on societies where its annual global cost reaches 400 billion dollars [8].
TBI is mainly classified according to severity into severe, moderate, and mild TBI, most commonly using the Glasgow Coma Scale (GCS) [9]. Mild TBI (mTBI), also known as concussion, affects about 740 per 100,000 people whereas severe TBI affects 73 per 100,000 people [2]. Post-concussive symptoms are regarded as transient and patients usually completely recover within three months [10]. Nevertheless, repetitive head traumas have been shown to have a grave long-term consequence such as the development of neurodegenerative diseases, including chronic traumatic encephalopathy (CTE) and Alzheimer’s diseases [11]. With the increasing worldwide attention towards sports, the increasing numbers of individuals enlisting in the military forces, and the overall increase in violence in societies, repetitive head trauma has resulted in as many as 1 million hospital admissions in 2018 [12].
The pathogenesis of TBI mainly comprises primary and secondary injuries. The primary injury is the direct result of the external physical force on the brain, whereas secondary injury happens minutes to days following the primary injury and consists of the molecular and chemical changes leading to neuronal damage [13]. However, the spatial separation between the primary and secondary injury is not clear in some forms of TBI, like repetitive mTBI, which shares some features with penetrating head TBI [14]. Ultimately, secondary damage can associate with behavioral, emotional, and cognitive deficits [15][16]. After the mechanical insult, there is a release of excitatory amino acids such as glutamate into the synapse that in turn overstimulate N-methyl-D-aspartate (NMDA) receptors. The resultant is an overload of Ca2+, along with increased depolarization due to ionic imbalance [17][18][19]. High levels of Ca2+ can cause an intracellular Ca2+-dependent Ca2+ release and subsequently activate Ca2+-dependent enzymes including proteases, lipases, and endonucleases that can eventually lead to protein degradation, disruption of protein phosphorylation, and protein aggregation like tau proteins [17]. The excess of intracellular Ca2+ and excitotoxicity lead to the excessive production of reactive oxygen species (ROS) and ultimately to oxidative stress. This primarily occurs in mitochondria where the increased Ca2+ stimulates ROS production through membrane transition pore (MTP) activation, cytochrome c (cyt c) release, and respiratory chain inhibition [20]. In addition, secondary injury post TBI also encompasses neuro-inflammation, blood brain barrier (BBB) dysfunction, axonal injury and metabolic disturbance.
2. Therapeutic Options Targeting Oxidative Stress in TBI
2.1. Edaravone
Edaravone is an antioxidant that has been used in Japan since 2001 in the management of neurological symptoms and functional disorders associated with acute ischemic stroke. The drug scavenges free radical post-ischemic events, thereby mitigating oxidative injury in neurons. Besides its anti-oxidative property, it has been shown to play a role in decreasing nitric oxide production, matrix metalloproteinases activity, inflammation, and apoptotic cell death, thus fitting into the class of multi-target compound [21]. Due to its promising effects, edaravone was launched as a therapeutic drug for amyotrophic lateral sclerosis (ALS) in Japan and Korea in 2015, and was later approved by U.S. FDA in 2017 and in Canada in 2018 [22].
2.1.1. Edaravone in ALS
The exact mechanism of action by which edaravone exerts its therapeutic effects in ALS is unknown; however, it may be due to its anti-oxidative property since oxidative stress is a part of the cascade leading to motor neuron death in patients with ALS [23]. In multiple in-vivo studies on ALS, edaravone was capable of suppressing the nitration of tyrosine residues, attenuating motor decline and muscle weakness, reducing the abnormal disposition of SOD1 in the spinal cord, preserving motor neurons, decreasing denervation atrophy, and decreasing motor neuron degeneration [24][25][26]. In vitro studies found that edaravone ameliorated the harmful effects of neurotoxins and oxidants in cultured neuronal cells by reducing ROS generation, decreasing cytotoxicity, and increasing cell survival. The mechanism underlying these neuroprotective effects was associated with the nuclear factor erythroid 2-related factor 2/anti-oxidant responsive element (Nrf2/ARE) pathway in which increased Nrf2 expression and translocation from the cytoplasm to the nucleus increased the expression of the antioxidant enzymes SOD and heme oxygenase-1 (HO-1) [27][28]. The effects of edaravone on mitochondrial dysfunction are poorly addressed; however, it was shown that edaravone improves mitochondrial functions and modulates mitochondrial-dependent apoptosis pathways by preventing cyt c release and inhibiting caspase-3 activation [29].
2.1.2. Edaravone in TBI
For the past decade, extensive research focused on the therapeutic effects of edaravone in several neurological disorders, including TBI, where it was shown to be safe and efficient. The novelty of this drug relies in its multi-target mechanism which makes it a drug of choice compared to other antioxidants. Edaravone was used in multiple animal models of TBI and resulted in enhanced molecular and behavioral outcomes. It was capable of ensuring neuroprotection by reducing injury volume, increasing neuronal number, and decreasing apoptosis [30][31][32]. An important role has been attributed to edaravone in fighting oxidative stress in TBI, where the total volume of nitrotyrosine (NT) and the production of ROS were suppressed following edaravone administration. Also, the expression of Nrf2, a transcription factor, and its downstream genes were elevated by edaravone [30]. Moreover, it can decrease H2O2 level and increase SOD and GPx levels [32]. Edaravone is also capable of attenuating the immune response by decreasing the production of the inflammatory cytokines, tumor necrosis factor α (TNFα), and interleukins 6 and 1-b (IL-6, IL-1b) and increasing IL-10, an anti-inflammatory cytokine. It can also reduce the expression of nuclear factor kappa B (NF‑κB) [31][32]. Edaravone was also reported to enhance the the production of stem cells, where a significant increase in nestin positive cells was noted around the injured brain area in at 3 and 7 days after TBI. Similarly, the number of isolated and cultured spheres was significantly increased [33]. Ultimately, edaravone treated animals can show modified behavior, by having lower neurological severity score (NSS) [32], and enhanced performance in beam-balancing and prehensile traction tests [31]. They also display improved learning and memory in Morris water maze [34] and lessened depressive-like behavior in the forced swim test [35].The mechanism and outcomes of edaravone are displayed in Figure 1.
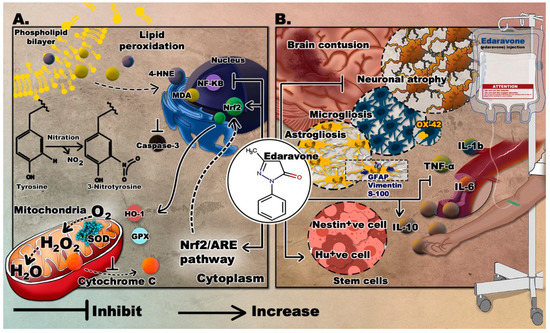
Figure 1. The proposed neurotherapeutic mechanisms of edaravone. (A) Suppression of oxidative stress by edaravone; (B) effects of edaravone in TBI. Molecular graphics of the superoxide dismutase (SOD) enzyme was visualized using the 3D Protein Imager online server [36].
2.2. Mitoquinone (MitoQ)
MitoQ is among the widely used antioxidants that target the mitochondria. It was developed in the 1990s to readily penetrate the BBB and neuronal membranes, where it is concentrated into several hundred-folds within the mitochondria where it mediates the local anti-oxidative capacity [37]. MitoQ is formed by covalently binding ubiquinone or coenzyme Q, an endogenous antioxidant and a component of the mitochondrial electron transport chain (ETC), to triphenylphosphonium (TPP+) ions. TPP+ is a lipophilic cation that drives the ubiquinone moiety into the inner mitochondrial membrane, by the negative electrochemical potential [38]. Within the ETC, complex II, also known as succinate dehydrogenase, reduces MitoQ ubiquinone moiety to the active antioxidant ubiquinol which scavenges excess ROS. After reducing ROS, ubiquinol is oxidized to ubiquinone and then recycled by complex II. MitoQ is a poor substrate for complexes I and III, so it cannot substitute for endogenous ubiquinone and therefore does not take part in the mitochondrial respiration; thus acting as a renewable antioxidant [39]. Notably, studies have shown that MitoQ produces ROS during its redox cycling [40]. This perturbation may trigger defense cascades to protect cells. MitoQ possibly induces the oxidation of Keap1 and its subsequent degradation and release of Nrf2 [41]. This eventually leads to the upregulated expression of antioxidant enzyme genes as mentioned previously. The formulation of MitoQ is summarized in Figure 3A.
2.2.1. Effects of MitoQ in Preclinical Studies of Neurodegenerative Diseases
In animal and in vitro models of PD, MitoQ has demonstrated positive outcomes. When used in SH-SY5Y cell line, MitoQ reduced 6-OHDA-induced mitochondrial fragmentation. It inhibited mitochondrial fission protein and the translocation of pro-apoptotic protein (Bax) in the mitochondria [42]. In another study, neuroprotective effects were demonstrated in both cellular and mouse models of PD, in which MitoQ treatment inhibited the loss of dopaminergic neurons and enhanced behavioral performance [43]. Furthermore, a recent study performed on a Zebrafish PD model showed that MitoQ could improve the oxidant-antioxidant balance, ameliorate the expressions of PD- related genes, and enhance the overall mitochondrial function [44].
Moreover, in a Huntington's disease (HD) mouse model, MitoQ treatment enhanced the fine motor control and reduced markers of oxidative damage in muscles. It also attenuated overactive autophagy-induction associated with muscle wasting [45]. Such intervention improved muscle performance and protected from proteostasis impairment, suggesting that mitochondrial-targeted antioxidants may have promising therapeutic effects in neuromuscular disorders.
Due to its promising effects in neuromuscular disorders, MitoQ was studied in several ALS-models. In transgenic ALS mice carrying the G93A mutated human SOD1, MitoQ treatment improved mitochondrial function in both the spinal cord and the quadriceps muscle [46]. Interestingly, the nitro-oxidative markers in the spinal cord of treated animals were significantly reduced, coupled with a recovery of neuromuscular junctions and an enhancement in hind-limb strength. MitoQ treatment also significantly prolonged the life span of SOD1 (G93A) mice.
In an Alzheimer’s disease (AD) mouse model, MitoQ treatment significantly improved spatial memory retention and reduced brain oxidative stress, astrogliosis, microglia cell proliferation, tau hyper-phosphorylation, and amyloid plaque formation [47]. MitoQ treatment also extended the lifespan of 3xTg-AD mice, thus emphasizing its role in dampening the neuropathology responsible for the accelerated death rate of these mice.
2.2.2. Effects of MitoQ in Preclinical Models of TBI
Evaluating the effects of MitoQ in a TBI model showed significant improvements in behavioral and molecular outcomes. Considering the fact that mitochondria play a crucial role in the pathogenesis of TBI, delivering a mitochondria-targeted antioxidant protects mitochondria against oxidative stress thereby preventing neuronal death. As discussed, mitochondria are a major source of ROS and hence are particularly sensitive to oxidative injury which further exacerbate ROS production. This can lead to a vicious cycle of an increasing level of mitochondrial injury and eventually apoptotic cell death and metabolic imbalance [48]. Accordingly, available research data indicate that decreased mitochondrial oxidative stress can suppress or delay the progression of TBI [49].
The first study to demonstrate the effects of MitoQ in TBI was recently completed. The treatment was shown to significantly improve neurological deficits, alleviate brain edema, and inhibit cortical neuronal apoptosis in a TBI mouse model [50]. Mice treated with 4mg/kg of MitoQ showed significantly improved neurobehavioral functions, coupled with increased activity of different antioxidant enzymes, inckuding SOD and GPx. The effect of MitoQ on TBI-induced apoptosis was evaluated by quantifying the expression of the mitochondrial apoptosis-related proteins. Bax protein translocation to the mitochondria and the cytosolic release of cyt-c were reduced, confirming that MitoQ can attenuate neuronal apoptosis in the cortical contusion post-injury. Remarkably, MitoQ accelerated the Nrf2 nuclear translocation and subsequently upregulated the expression of downstream proteins, including HO-1 and quinone oxidoreductase 1. Therefore, such findings demonstrate that MitoQ mediates its neuroprotective effects via activating the Nrf2/ARE pathway. The role of mitoQ in apoptosis is depicted in Figure 3B.
Considering its potential effects in preclinical studies of several neurological diseases, MitoQ intervention can be a promising drug to attenuate the progression of TBI, especially that oxidative stress and mitochondrial impairment play a significant role in the pathogenesis of TBI. The proposed role of MitoQ in TBI can be found in Figure 3C.
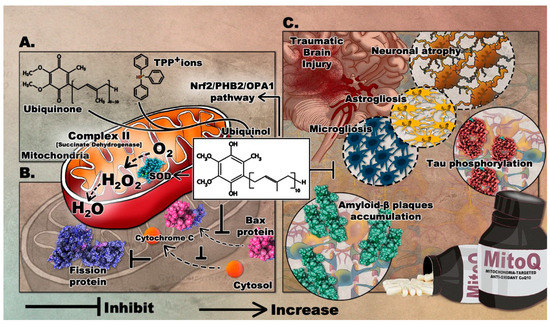
Figure 3. The mediated therapeutic mechanisms of MitoQ targeting the mitochondria with its proposed implications on TBI. (A) Formation of MitoQ; (B) role of MitoQ in attenuation of neuronal apoptosis; (C) inhibitory role of MitoQ following TBI. Molecular graphics of the Superoxide Dismutase (SOD) enzyme, fission protein, Bax protein, Amyloid-ß plaques, and phosphorylated Tau were visualized using the 3D Protein Imager online server [36].
3. Conclusion
Antioxidants such as MitoQ and edaravone were shown to have a neuroprotective effect in several pre-clinical studies. Such outcomes include enhanced neurological and motor functions, as well as reduced oxidative stress, neuroinflammation, and apoptosis; i.e., alleviation of TBI hallmarks. Treating TBI remains a necessity considering its growing impact on societies. Despite the potential role of antioxidants, further studies are needed to elucidate the mechanisms by which antioxidants work and to find ways to enhance their mode of delivery. Furthermore, clinical studies are also needed to prove the efficacy of antioxidants as they move from pre-clinical settings to bedside. Hence, drug repurposing could be a promising initiative in managing TBI.
References
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640.
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1–18.
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696.
- Phillips, S.; Woessner, D. Sports-related traumatic brain injury. Prim. Care 2015, 42, 243–248.
- Wojcik, B.E.; Stein, C.R.; Bagg, K.; Humphrey, R.J.; Orosco, J. Traumatic brain injury hospitalizations of U.S. army soldiers deployed to Afghanistan and Iraq. Am. J. Prev. Med. 2010, 38, S108–S116.
- Johnson, W.D.; Griswold, D.P. Traumatic brain injury: A global challenge. Lancet Neurol. 2017, 16, 949–950.
- El-Menyar, A.; Mekkodathil, A.; Al-Thani, H.; Consunji, R.; Latifi, R. Incidence, demographics, and outcome of traumatic brain injury in the Middle East: A systematic review. World Neurosurg. 2017, 107, 6–21.
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048.
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet (Lond. Engl.) 1974, 2, 81–84.
- McCrea, M.; Guskiewicz, K.M.; Marshall, S.W.; Barr, W.; Randolph, C.; Cantu, R.C.; Onate, J.A.; Yang, J.; Kelly, J.P. Acute effects and recovery time following concussion in collegiate football players: The NCAA Concussion Study. JAMA 2003, 290, 2556–2563.
- Yang, Z.; Lin, F.; Weissman, A.S.; Jaalouk, E.; Xue, Q.S.; Wang, K.K. A repetitive concussive head injury model in mice. J. Vis. Exp. 2016, 116, 54530.
- Pervez, M.; Kitagawa, R.S.; Chang, T.R. Definition of traumatic brain injury, neurosurgery, trauma orthopedics, neuroimaging, psychology, and psychiatry in mild traumatic brain injury. Neuroimaging Clin. N. Am. 2018, 28, 1–13.
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic brain injury: Current treatment strategies and future endeavors. Cell Transplant. 2017, 26, 1118–1130.
- Fehily, B.; Fitzgerald, M. Repeated mild traumatic brain injury: Potential mechanisms of damage. Cell Transplant. 2017, 26, 1131–1155.
- Albensi, B.C. Models of brain injury and alterations in synaptic plasticity. J. Neurosci. Res. 2001, 65, 279–283.
- Zohar, O.; Schreiber, S.; Getslev, V.; Schwartz, J.P.; Mullins, P.G.; Pick, C.G. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 2003, 118, 949–955.
- Barkhoudarian, G.; Hovda, D.A.; Giza, C.C. The molecular pathophysiology of concussive brain injury—An update. Phys. Med. Rehabil. Clin. N. Am. 2016, 27, 373–393.
- Hayes, R.L.; Jenkins, L.W.; Lyeth, B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: Acetylcholine and excitatory amino acids. J. Neurotrauma 1992, 9 (Suppl. S1), S173–S187.
- Strecker, G.J.; Jackson, M.B.; Dudek, F.E. Blockade of NMDA-activated channels by magnesium in the immature rat hippocampus. J. Neurophysiol. 1994, 72, 1538–1548.
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264.
- Kikuchi, K.; Kawahara, K.-I.; Uchikado, H.; Miyagi, N.; Kuramoto, T.; Miyagi, T.; Morimoto, Y.; Ito, T.; Tancharoen, S.; Miura, N.; et al. Potential of edaravone for neuroprotection in neurologic diseases that do not involve cerebral infarction. Exp. Ther. Med. 2011, 2, 771–775.
- Yoshino, H. Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev. Neurother. 2019, 19, 185–193.
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299.
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455.
- Aoki, M.; Warita, H.; Mizuno, H.; Suzuki, N.; Yuki, S.; Itoyama, Y. Feasibility study for functional test battery of SOD transgenic rat (H46R) and evaluation of edaravone, a free radical scavenger. Brain Res. 2011, 1382, 321–325.
- Ikeda, K.; Iwasaki, Y. Edaravone, a free radical scavenger, delayed symptomatic and pathological progression of motor neuron disease in the wobbler mouse. PLoS ONE 2015, 10, e0140316.
- Shou, L.; Bei, Y.; Song, Y.; Wang, L.; Ai, L.; Yan, Q.; He, W. Nrf2 mediates the protective effect of edaravone after chlorpyrifos-induced nervous system toxicity. Environ. Toxicol. 2019, 34, 626–633.
- Zhang, L.; Guo, Y.; Wang, H.; Zhao, L.; Ma, Z.; Li, T.; Liu, J.; Sun, M.; Jian, Y.; Yao, L.; et al. Edaravone reduces Abeta-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci. 2019, 221, 259–266.
- Li, B.; Yu, D.; Xu, Z. Edaravone prevents neurotoxicity of mutant L166P DJ-1 in Parkinson’s disease. J. Mol. Neurosci. 2013, 51, 539–549.
- Miyamoto, K.; Ohtaki, H.; Dohi, K.; Tsumuraya, T.; Song, D.; Kiriyama, K.; Satoh, K.; Shimizu, A.; Aruga, T.; Shioda, S. Therapeutic time window for edaravone treatment of traumatic brain injury in mice. Biomed. Res. Int. 2013, 2013, 379206.
- Wang, G.H.; Jiang, Z.L.; Li, Y.C.; Li, X.; Shi, H.; Gao, Y.Q.; Vosler, P.S.; Chen, J. Free-radical scavenger edaravone treatment confers neuroprotection against traumatic brain injury in rats. J. Neurotrauma 2011, 28, 2123–2134.
- Zhang, M.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Xiao, J.; Zhang, H.Y.; Chen, D.Q. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp. Ther. Med. 2019, 18, 467–474.
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, H. The novel free radical scavenger, edaravone, increases neural stem cell number around the area of damage following rat traumatic brain injury. Neurotox. Res. 2009, 16, 378–389.
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Imano, M.; Hashimoto, S.; Ito, H. Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neurochem. Res. 2010, 35, 348–355.
- Higashi, Y.; Hoshijima, M.; Yawata, T.; Nobumoto, A.; Tsuda, M.; Shimizu, T.; Saito, M.; Ueba, T. Suppression of oxidative stress and 5-lipoxygenase activation by edaravone improves depressive-like behavior after concussion. J. Neurotrauma 2014, 31, 1689–1699.
- Tomasello, G.; Armenia, I.; Molla, G. The protein imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911.
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 629–656.
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412.
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312.
- Doughan, A.K.; Dikalov, S.I. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal. 2007, 9, 1825–1836.
- Rao, V.A.; Klein, S.R.; Bonar, S.J.; Zielonka, J.; Mizuno, N.; Dickey, J.S.; Keller, P.W.; Joseph, J.; Kalyanaraman, B.; Shacter, E. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 2010, 285, 34447–34459.
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Jiménez, M.D.M.A.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182.
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684.
- Ünal, İ.; Çalışkan-Ak, E.; Üstündağ, Ü.V.; Ateş, P.S.; Alturfan, A.A.; Altinoz, M.A.; Elmaci, I.; Emekli-Alturfan, E. Neuroprotective effects of mitoquinone and oleandrin on Parkinson’s disease model in zebrafish. Int. J. Neurosci. 2019, 130, 1–9.
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382.
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodriguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213.
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409.
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380.
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531.
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899.

