Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Darina Bačenková | -- | 4575 | 2022-08-16 17:50:49 | | | |
| 2 | Rita Xu | -4 word(s) | 4571 | 2022-08-17 03:44:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bačenková, D.; Trebuňová, M.; Morochovič, R.; Dosedla, E.; Balogová, A.F.; Gašparová, P.; Živčák, J. Mesenchymal Stem Cells in Rheumatoid Arthritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/26210 (accessed on 23 June 2026).
Bačenková D, Trebuňová M, Morochovič R, Dosedla E, Balogová AF, Gašparová P, et al. Mesenchymal Stem Cells in Rheumatoid Arthritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/26210. Accessed June 23, 2026.
Bačenková, Darina, Marianna Trebuňová, Radoslav Morochovič, Erik Dosedla, Alena Findrik Balogová, Petra Gašparová, Jozef Živčák. "Mesenchymal Stem Cells in Rheumatoid Arthritis" Encyclopedia, https://encyclopedia.pub/entry/26210 (accessed June 23, 2026).
Bačenková, D., Trebuňová, M., Morochovič, R., Dosedla, E., Balogová, A.F., Gašparová, P., & Živčák, J. (2022, August 16). Mesenchymal Stem Cells in Rheumatoid Arthritis. In Encyclopedia. https://encyclopedia.pub/entry/26210
Bačenková, Darina, et al. "Mesenchymal Stem Cells in Rheumatoid Arthritis." Encyclopedia. Web. 16 August, 2022.
Copy Citation
Rheumatoid arthritis (RA) is an autoimmune disease that causes damage to joints. The possibility of influencing the disease through immunomodulation by mesenchymal stem cells (MSCs). There is an occurrence of rheumatoid factor and RA-specific autoantibodies to citrullinated proteins in most patients. Citrulline proteins have been identified in the joints of RA patients, and are considered to be the most suitable candidates for the stimulation of anti-citrulline protein antibodies production.
mesenchymal stem cells
rheumatoid arthritis
immunomodulation
1. Introduction
Rheumatoid arthritis (RA) is a chronic systemic disease that causes damage to joints, connective tissue, muscles and tendons [1]. Chronic inflammation is a hallmark of RA joint pathology synovitis, which causes cartilage and bone tissue erosion by resident synoviocyte-like fibroblasts (FLS). Joint inflammation is initiated and maintained by autoimmune mechanisms [2]. The preclinical phase of RA, which takes a long period of time before the first clinical symptoms appear, is characterized by the presence of circulating autoantibodies, and increased levels of inflammatory cytokines and chemokines. In RA, inner lining hyperplasia occurs and the numbers of macrophages and fibroblast-like synoviocytes (FLSs) are elevated (Table 1). Inflammatory cells penetrate to the rheumatoid joint, where they are activated and contribute to local destruction. More specifically, they are mainly neutrophils that accumulate in the synovial fluid, absorb immune complexes, and release proteolytic enzymes [3].
Table 1. Abbreviations often used in the text.
| Abbreviations | Acronym |
|---|---|
| Anti-citrullinated protein antibodies | ACPAs |
| C-X-C chemokine receptor | CXCR |
| Dendritic cells | DC |
| Extracellular matrix | ECM |
| Extracellular vesicles | EVs |
| Fibroblast-like synoviocytes | FLSs |
| Immunoglobulins | Ig |
| Indolamine 2,3-dioxygenase | IDO |
| Interferon gamma | IFN-γ |
| Interleukin | IL |
| Intracellular adhesion molecule 1 | ICAM1 |
| Macrophage inflammatory proteins | MIP |
| Matrix metalloproteinases | MMP |
| Mesenchymal stem cells | MSCs |
| Natural killer cells | NK cells |
| Peptidyl arginine deiminase | PAD |
| Prostaglandin E2 | PGE2 |
| Rheumatoid arthritis | RA |
| Synovial fluid | SF |
| T regulatory cells | Treg |
| Toll-like receptors | TLR |
| Transforming growth factor beta | TGF-β |
| Tumor necrosis factor alpha | TNF-α |
| Vascular adhesion molecule 1 | VCAM1 |
Inflammation in the synovium is accompanied by the formation of a pannus in the joint lining. A pannus is an aggressive front tissue that destroys the local joint structures. Panus synoviocytes produce microscopic structures called lysosomes that release enzymes (proteins) called matrix metalloproteinases (MMPs), which have a degrading effect on cartilage. Altered cell metabolism ultimately leads to joint erosion and damage. The exact cause of RA is currently not fully understood. It is most likely to be a disease that occurs in genetically predisposed individuals and under certain circumstances, for example, as an acute disease in the presence of pathogenic microorganisms. RA disease is characterized by chronic inflammation, which is maintained by autoimmune processes. The course of RA is highly variable. Overall, however, the course is progressive and often leads to disability. The goal of treatment is to achieve remission, but the treatment must be timely and aggressive [1][2][3]. RA treatment is aimed slowing the progression of the disease by inhibiting the inflammation of the affected parts. Currently applied treatment methods include synthetic and disease-modifying anti-rheumatic drugs (DMARDs), non-steroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids. Several side effects are present in patients following the glucocorticoids’ administration. A higher dose of glucocorticoids causes parchment skin, leg edema, sleep disorders, immunosuppression, weight gain, hypertension and diabetes. Recently, antirheumatic drugs of the type DMARDs, and especially the DMARD methotrexate (MTX), have been used therapeutically; furthermore, new biological substances have improved the treatment of RA. Interestingly, early treatment more effectively increases its effectiveness and reduces the extent of joint damage. However, there is a certain problem with catching and treating patients in the early stages of the disease; NSAIDs are indicated as therapy in combination with other medications during the RA phase. This is mainly a symptomatic treatment. Undesirable side effects are common with this therapy. DMARDs objectively suppress the inflammatory reaction, including the inhibition of the immediate phase reaction, and also reduce damage to the joint, which has been confirmed by X-ray analysis. The most frequently used drugs are sulfasalazine and methotrexate. Biological therapy is a modern alternative to RA therapy. In principle, it involves blocking the activity of the main inflammatory cytokine, tumor necrosis factor alpha (TNF-α). There are several preparations based on the neutralizing antibodies against TNF-α. Biological therapy is recommended for patients with a severe form of RA, mainly for patients who respond poorly to DMARDs treatment. Undesirable effects may occur, such as infections, tuberculosis, and malignant diseases. Biologic DMARDs and NSAIDs, which are known as effective painkillers, as well as other expensive drugs with a high potential to reduce disease symptoms and prevent disease progression in patients, are most commonly used in RA therapy. Cytokines such as TNF-α, Interleukin 1 (IL-1), IL-6, IL-7, IL-15, IL-17, IL-18, IL-21, IL-23, IL-32, IL-33 and granulocyte-macrophage colony stimulating factor (GM-CSF) are important in the development of RA. Interestingly, the results of clinical studies that focused on blocking IL-1, IL-17 and IL-18 did not show significant success. In contrast, blocking TNF-α or IL-6 was successful in influencing symptoms [4]. The negatives of RA treatment are its serious side effects. Moreover, treatment is not effective for all patients. In RA, up to 30% of patients do not respond to conventional treatment [5].
1.1. The Immunology of Rheumatoid Arthritis
Autoimmune diseases are manifested as disturbances in the inflammatory process, while the affected area is disturbed and its functionality is lost in the case of neglecting the necessary therapy. Changes in the joint during the RA process are related to an unbalanced biochemical process of catabolism and anabolism, while structural disorders occur in the joint. These serious biochemical changes that occur in connection with inflammatory synovitis in RA have the effect of suppressing the natural remodeling of structures in the joint. RA disease is characterized by the presence of autoantibodies in the period before the manifestation of the disease itself [6]. The occurrence of anticitrulline antibodies is related to the formation of immune complexes in the affected joint. The course of the autoimmune disease RA involves changes in both innate and adaptive immunity in subsets of T-lymphocytes, the cells of the monocyte, and the macrophage lineage. Among the manifestations of RA is a decrease in immune tolerance, which includes decreased T-reg cells. On the contrary, inflammatory cells and their activation increase, which is also related to the increased production of cytokines, which ends in chronic inflammation. The pro-inflammatory cytokines IL-6 and TNF-α are present at the site of inflammation. Due to the extent of the disease and the multiple affected parts, the disease is perceived as a systemic disease. Synovial hyperplasia is a very common manifestation of RA after joint injuries, and the stroma of the synovial tissue contain cells with a characteristic MSC phenotype. According to the available data, it has been demonstrated that MSCs can migrate from the BM into the joint cavity, and they have been detected in the proliferating synovial tissue. Authors de Bari et al. demonstrated the presence of MSCs in the human synovial membrane which have a characteristic phenotype and differentiation ability. MSCs, as stem cells, have an important role in homeostasis, while in the inflammatory process this balance is dysregulated [4][5].
1.2. Autoantibodies in Rheumatoid Arthritis
Monitoring the presence of autoantibodies in patients, rheumatoid factor (RF) and the autoantibodies to post-translationally modified proteins such as citrullination (anticitrulin antibodies) are now increasingly important in the diagnosis of disease. Autoantibodies develop in the serum and synovial fluid (SF) of patients with RA. Antibodies cause the formation of immune complexes in the joints, which results in the activation of immune cells through the complement system, as well as the direct activation of immune cells and the secretion of chemokines and cytokines that enhance the immune response. RF antibodies that recognize the Fc-terminus of immunoglobulins (Ig)-Gs are the standard and first type of autoantibodies found in RA. Now, it is known that the RF response uses a wide-range spectrum of isotypes, including IgM, IgG and IgA. The RF antibody is present in about 75% of RA patients, and has also been determined in other diseases and non-rheumatic conditions such as leprosy, syphilis, pulmonary tuberculosis, chronic liver disease, and sarcoidosis, as well as in many rheumatological diseases such as systemic lupus erythematosus (SLE) and Sjogren’s disease. In the current infection, RF can act to bind IgG-coated pathogens and condition their removal in the form of immune complexes [6]. Genetic factors have been shown to account for about 60% of the risk of developing RA. Anti-citrulline protein antibodies (ACPA) and carbamylation (anti-CarP antibodies) are very important. A risk factor for ACPA-positive RA is the HLA class II molecule HLA-DRB1-“shared epitope” (SE) alleles. Recent studies suggest that the HLA-DRB1 alleles, which encode SE, a five-amino acid sequence motif at residues 70–74 of the HLA-DRβ chain, are significantly present in severe RA. Another interesting assumption is the refinement of the SE motif, which relates to position 70 of the DRβ chain. Current facts suggest that while glutamine or arginine at position 70 appears to be a significant risk for RA, aspartic acid at this position provides protection [7]. The risk is much higher for people carrying one SE or two SE alleles compared to SE-negative individuals [8]. The incidence of RA by gender is higher in women compared to men, and the incidence rate in women is more than double. Due to a higher prevalence in women, reproductive and hormonal factors are thought to affect the disease. The exact role of gonadotropins, estrogens and androgens in RA is currently unknown. Estrogens are thought to be more pro-inflammatory and androgens more anti-inflammatory. The results of the studies are not yet clear [9]. There is a form of seronegative RA that is characterized by the absence of the autoantibodies RF and ACPA in serum. In addition, there are several differences in etiopathogenesis and different risk factors. Several authors have summarized data from studies that describe a non-seropositive form of RA. The RA treatment regimen does not currently take into account the presence or absence of autoantibodies in patients, and the therapy is largely identical [10].
1.3. Post-Translational Modifications of Proteins ECM
Specific serological markers of RA are antibodies to citrullinated proteins, which show higher specificity in RA compared to RF [11][12]. ACPAs specifically bind to citrullination after translational modification (PTM). ACPAs are synthesized by immune cells in the panus; thus, the trigger for ACPA production is the autoantigen present in the affected joint. Citrullination has been found in several body parts, as well as in both mouse and human inflamed joints. It is a PTM that can alter the interaction properties of proteins that contain lysine and arginine in their interactive domains. As previously mentioned, the post-translational process of citrullination, which is driven by calcium and the enzyme peptidyl arginine deiminase (PAD), leads to the removal of a positive charge for each arginine residue that is converted to neutral citrulline [13]. Intracellular enzyme PAD is an evolutionarily conserved protein with several isoforms in both mice and humans (PAD1-4 and PAD6). While PAD4 is present in monocytes and macrophages, both PAD2 and PAD4 are characteristic of SF. Both apoptosis and necrosis are associated with a locally increased calcium concentration, which also releases PAD. It is thought that even in inflamed tissues, PAD could act on the citrullination of ECM proteins such as fibrinogen and collagen. Citrulline collagen II, α-enolase, fibrinogen and vimentin have been detected in the joints of patients with RA, and are considered to be the most suitable candidates for the stimulation of ACPA production [14]. In donors who provided serum years before their first RA symptoms appeared, the ACPA test sensitivity was about 50%. Interestingly, ACPA autoantibody serums were detectable in several patients aged 10–20 years before the onset of the disease [15]. Post-translational changes in fibronectin and collagen proteins that are part of the extracellular matrix (ECM) can potentially affect cell properties such as adhesion, physicochemical properties and antigenicity [16][17]. One of the multifactorial causes of RA is associated with dysregulated ECM remodeling, which is an interesting fact for citrullinated vimentin. Citrullination, which has a negative effect on the assembly of vimentin filaments, leads to the increased formation of soluble precursors that are transported extracellularly. The extracellular localization of these precursors subsequently induces autoimmune responses in joints with RA. Citrullinated vimentin expression increases following the cell damage. It is also strongly expressed in the repair modulating cells, which stimulates their migration. The local production of posttranslational modified vimentin in the synovium of RA patients is expected. This potential mechanism of posttranslational modified vimentin (PTMV) formation could be related to inflammation. Macrophages and neutrophils that migrate from the periphery have high levels of PAD-2 and PAD-4 [18]. Vimentin is most commonly localized in the cytoplasm and nucleus, and to a lesser extent on the cell surface in the form of post-translationally modified phosphorylated vimentin. The endoplasmic reticulum and Golgi complex are involved in the process of vimentin formation, forming vesicles that are secreted extracellularly as phosphorylated vimentin. The cytokines present may regulate the expression of phosphorylated vimentin on the cell surface. Interestingly, MSCs produced by TNF-α and IL-10 reduce vimentin protein exocytosis. The exact physiological function of citrullination is not fully understood; however, it is known to cause the increased susceptibility of proteins to proteolytic degradation. Another interesting finding is that these proteins are processed antigen-present cells (APCs), which can be citrullinated prior to their presentation to T cells [19][20]. It is also interesting to point out that there is an involvement of citrullinated proteins in the pathogenic mechanism of RA in animal models. Hill et al. described the induction of RA in a transgenic mouse with the HLA-DR4-IE gene by citrulline fibrin, which is commonly found in inflamed synovial tissue, and is also a common target of autoantibodies in RA patients [21]. RA disease in antigen-induced mice was characterized by synovial hyperplasia. Furthermore, citrullination has been found to induce immunological intolerance for endogenous reasons. Citrullinated proteins significantly induced synovial arthritis in animals. Higher numbers of CD4+ T cell were recorded in the synovium of RA animals. T cells altered by antigen-presenting cells expressing citrullinated peptides appear to be a putative cause of T cell-dependent ACPA production and joint damage [22] (Figure 1).
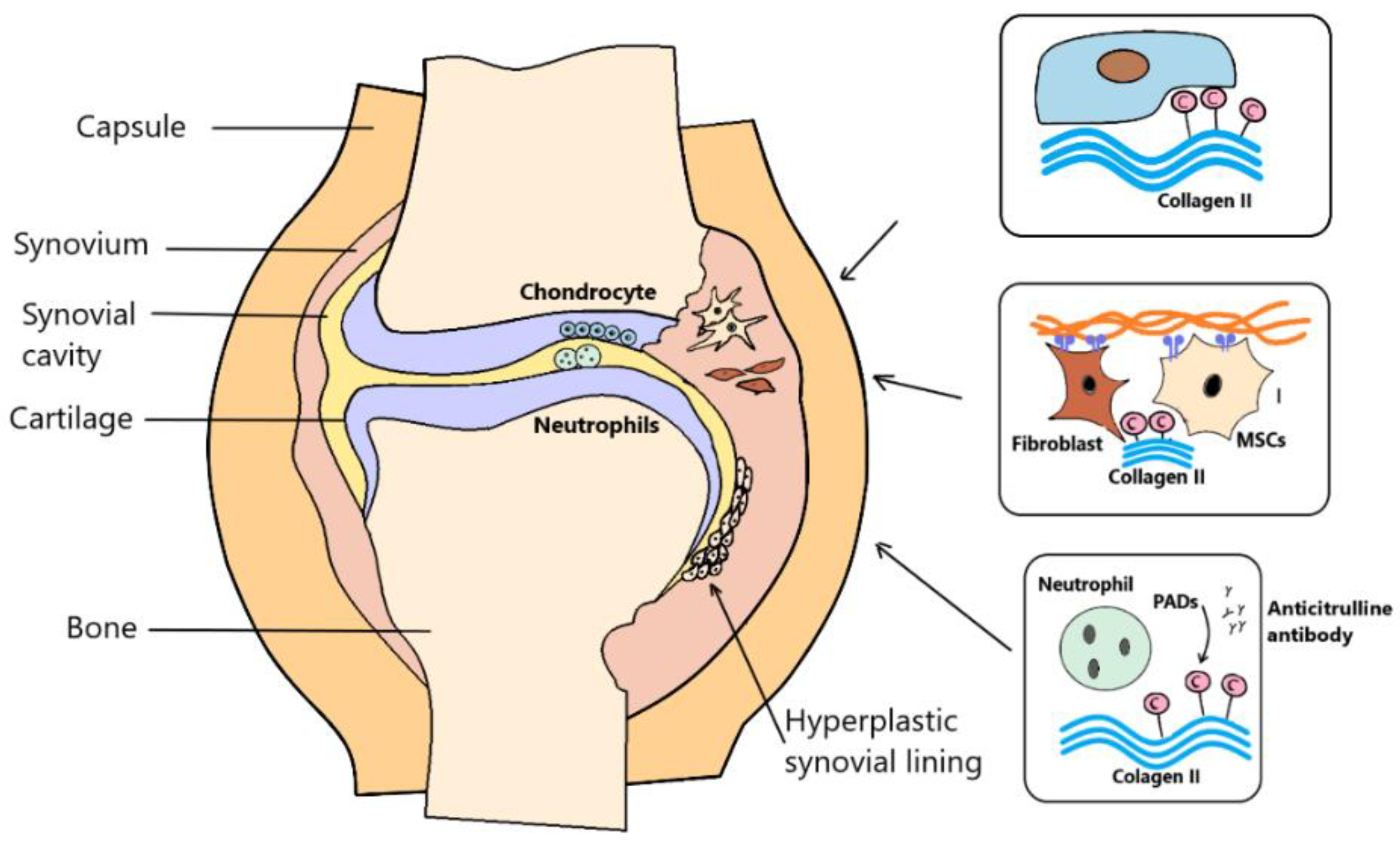
Figure 1. Antibodies to citrullinated proteins are specific serological markers of rheumatoid arthritis (RA). Anti-citrulline protein antibodies (ACPA) specifically bind to citrullination after translational modification. ACPAs are synthesized by immune cells in the panus, such that the trigger for ACPA production is the autoantigen present in the affected joint. Peptidyl arginine deiminase (PAD) are characteristic of synovial fluid. In inflamed tissues, PAD could act on the citrullination of extracellular matrix (ECM) proteins such as fibrinogen and collagen. Citrulline collagen II, fibrinogen and vimentin were found in the joints of RA patients.
2. Mesenchymal Stem Cells
Progress in cell therapy research has now been made, and the possibilities of regenerative procedures in patients with autoimmune diseases are being intensively investigated. Mesenchymal stem cell (MSC)-based therapy is a new alternative to the standard treatment of autoimmune diseases. It has been shown that several features of MSCs are suitable for the treatment. In the last century, MSCs were identified to be part of the bone marrow stroma as heterogeneous cell populations. They are capable of self-renewal and tissue regeneration, and have strong immunosuppressive properties [23]. MSCs are relatively easy to isolate from different parts of the organism, and are therefore ideal for in vivo and in vitro testing. MSCs have been described by Friedenstein as adherent, fibroblast-like cells with high self-renewal capacity [24]. Another characteristic feature of MSCs is their multilineage differentiation ability in vitro and in vivo. In particular, differentiation into multiple cell types, osteoblasts, chondrocytes, adipocytes, myocytes, epithelial cells and cardiomyocytes has been described [25][26][27][28][29][30][31]. Due to the absence of a unique marker, the MSC phenotype is defined by the expression of markers CD105, CD73, and CD90, and at the same time by the absence of the expression of the hematopoietic markers CD45, CD34, CD14, CD11b, CD79a, and CD19, and the MHC II molecule class [32] (Table 2).
Table 2. Phenotype of mesenchymal stem cells.
| Positive ≥ 95% | Negative ≤ 2% |
|---|---|
| CD105 | CD34 |
| CD73 | CD14, CD11b |
| CD90 | CD79 alpha, CD19 |
| HLA-DR |
Friendenstein was the first to describe the growth of so-called “colony-forming units—fibroblasts” (CFU-F) in bone marrow cultivation. The ability to form colonies still remains an important test for the quality of the examined cell sample. CFU-F is an indicator of the in vitro sample quality, recording the frequency of MSCs [33]. Bone marrow is the traditional source of MSCs. Another preferred source of MSCs is adipose tissue [34]. The isolation of MSCs is possible from many parts of the body’s organs and tissues. A promising source of MSCs is the chorion, part of the amniotic sac and the placenta [35][36][37]. The placenta is a rich source that contains two types of MSCs: amniotic mesenchymal stromal cells (AMSCs) and chorionic mesenchymal stromal cells (CMSCs). CMSCs can be isolated from the chorionic mesoderm layer [38][39][40]. CMSCs express the embryonic markers octamer-binding protein 4 (OCT4), SRY-related HMG-box 2 (Sox-2), homeoprotein Nanog, stage-specific embryonic antigen-4 (SSEA-4), and GATA binding protein 4 (GATA-4) [41] (Table 3). Overall, the main criteria for defining MSCs are as follows: cell adhesion to plastic, a specific surface antigen expression pattern, and specific differentiation potential towards osteogenic, adipogenic and chondrogenic lineages. The ability of MSCs to differentiate into cartilage, which is present in joint tissues, promotes therapeutic interventions by replacing damaged cells [42].
Table 3. The surface markers and identity of human mesenchymal stem cells.
| Markers | Human MCSs | Properties/Functions |
|---|---|---|
| CD105/Endoglin | Bone marrow MSCs (BM MSCs), adipose tissue MSCs (ADSC), umbilical blood cord MSCs (UCB MSCs) | A type I transmembrane protein reported to induce activation and proliferation of endothelial cells and co-receptor for (transforming growth factor beta) TGF-β [34] |
| CD90/Thy-1 | BM MSCs, ADSCs, UCB MSCs | Surface marker hypothesized to function in cell-cell and cell-matrix interactions, nerve regeneration, apoptosis, inflammation [32] |
| CD73/Ecto-5′-nucleotidase | BM MSCs, ADSC, UCB MSCs | Catalyzes the conversion at neutral pH of purine 5-prime mononucleotides to nucleosides, the preferred substrate being adenosine 5′-monophosphate (AMP) [32] |
| Stro-1 | BM MSCs, Amnion MSCs (AMSCs), Synovial membrane derived MSCs | Cell surface antigen in human bone marrow cells capable of differentiating stromal cells with a vascular smooth muscle-like phenotype, adipocytes, osteoblasts and chondrocytes [35] |
| CD271/LNGFR/Low-affinity nerve growth factor receptor | BM MSCs, ADSC, Placenta MSCs, Wharton Jely derived MSCs (WJ MSCs), AMSCs, Chorion MSCs (CMSCs) | The specific markers for the purification of human BM-MSCs [36] |
| Oct-4/Octamer-binding protein 4 | AMSCs, CMSCs | Transcription factors for pluripotency and self-renewal [35] |
| SSEA-4/Stage-specific embryonic antigen-4 | BM MSCs, Synovial membrane derived MSCs | Stage-specific embryonic antigen and MSCs from whole human bone marrow [35][41] |
| CD146/MCAM/Melanoma cell adhesion molecule | BM MSCs, Synovial membrane derived MSCs, Pericytes | Receptor for laminin alpha 4, a matrix molecule that is broadly expressed within the vascular wall [37] |
| Sox11/SRY-Box Transcription Factor 11 | BM MSCs | Marker downregulated during culture. Knockdown affect proliferation and osteogenesis potential [41] |
| CD349/Frizzled-9 | AMSCs, CMSCs | A novel marker for isolation of MSC from placenta. Members of the ‘frizzled’ gene family encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins [35] |
2.1. Mesenchymal Stem Cells in Synovium, in Their Native Environment
Synovium is an important tissue in RA, and its membrane lines the synovial joint cavity. Synovium has a lubricating effect on articular surfaces and provides nutrition to articular cartilage. Cells that exhibit the MSC phenotype can be isolated from synovium, where they are located mainly in the lining niche and the sublining perivascular niche. They may function as a reservoir of stem cells to repair joint structures, such as articular cartilage, with a very limited repair potential. In addition to MSCs, FLSs are an important part of the synovial membrane. In vitro, it is possible to isolate a mononuclear fraction of cells from a synovial membrane that contains both FLS and MSC. It is currently thought that MSCs may be involved in the anti-inflammatory processes of RA. In certain circumstances, the pro-inflammatory effects of MSCs are not ruled out. In an inflammatory environment, the MSC populations may be affected by inflammatory cytokines. It may be assumed that MSCs might have similar—but not exactly the same—functions. Synovial MSCs have been described as phenotypically heterogeneous, with similar populations of MSCs. An interesting fact is that the number of MSCs with in vitro multiline potential was significantly lower in the SF of RA patients compared to patients with osteoarthritis [43].
The effects of the cytokines present in the RA process on cell populations were observed experimentally. TNF-α prevents the differentiation ability of MSCs on the chondogenic line in vitro, and thus may contribute to the reduced ability of MSCs to repair joints and regenerate cartilage and bone during RA. Interferon gamma (IFN-γ) acts on the MSC as an activator of immunosuppressive and pro-inflammatory properties at the site of inflammation. Inflammation that occurs after tissue damage is usually accompanied by the infiltration of macrophages and neutrophils. This process is associated with phagocytosis by a macrophage, and is also accompanied by the release of pro-inflammatory factors IFN-γ, TNF-α, and IL-1. Furthermore, the body’s response to damage affects adaptive immunity and causes the activation of CD4+ and CD8+ T cells and B cells. The immunomodulatory role of MSC pro-inflammatory cytokines—IFN-γ, TNF-α and IL-1—can stimulate the immunosuppressive abilities of MSCs in a process that has been named “licensing”. Licensed MSCs with increased immunomodulatory capacity produce anti-inflammatory cytokines [6]. In an in vitro culture of FLS and MSCs, the transcription factor nuclear factor-κB is present. It acts to inhibit the differentiation capacity of MSCs, and at the same time stimulates ECM degradation processes [44]. Inflammatory processes in the joint affect the phenotype of cell populations, and an effect on the phenotype of FLSs and MSCs cells is expected. These populations may support further disease progression. It can be assumed that FLSs can also circulate and attack unaffected joints [45].
2.2. MSCs and Immune Cells in the Inflammatory Environment of Damaged Tissue
Chondroprogenitors have been described in areas of cartilage damage due to the inflammatory process. These could also originate from differentiated MSCs from the bone marrow. It can be assumed that MSCs migrate locally in the joint, and that reparative processes are limited, which mainly concerns the cartilage. MSCs with proven multilineage mesodermal potential can influence the disturbed imbalanced synovial environment. High levels of inflammatory cytokines in RA can also have a suppressive effect on stem cells. TNF-α suppresses the multilineage differentiation capacity of MSCs in vitro. TNF-α has a catabolic effect on cartilage and bone tissue. In patients with RA, the incidence of MSCs in synovial fluid is lower compared to MSCs in a healthy control group. MSCs have been found to have some pro-inflammatory properties in chronic inflammatory environments [14]. Inflammation that occurs after tissue damage is usually accompanied by the infiltration of macrophages and neutrophils. This process is associated with phagocytosis by a macrophage, and is also accompanied by the release of pro-inflammatory factors. Furthermore, the body’s response to damage affects adaptive immunity and causes the activation of CD4+ and CD8+ T cells and B cells. MSCs secrete several cytokines that have a direct effect on damaged tissue and a positive paracrine effect on the repair. MSCs are considered stem cells that cooperate in the repair of damage and are able to migrate to the site of injury. At the site of the defect, they cooperate with several types of inflammatory cells. According to the available data, MSCs are capable of secreting many cytokines and growth factors. Several of the mentioned factors are generated upon the principle activation of factor-κB (NF-κB) after exposure to pro-inflammatory stimuli such as IFN-γ, TNF-α, IL-1β, and lipopolysaccharide (LPS). These are TGF-β, FGF, vascular endothelial growth factor (vEGF), platelet-derived growth factor (PDGF), insulin-like growth factor 1 (IGF-1), and stromal cell-derived factor 1 (SDF-1) [23].
2.2.1. Homing and Migration of MSCs
An environment with a higher level of cytokines stimulates the differentiation of progenitor cells into fibroblasts and endothelial cells, which directly participate in tissue repair. In addition, in MSCs there is an adaptation in the expression of molecules such as intracellular ICAM-1, VCAM-1, and galectins. MSC-mediated therapy is conditioned by the ability of MSCs to home in on and migrate to the site of the injury. MSCs have the ability to attach to endothelial cells or ECM proteins such as collagen, and fibronectin and laminin thanks to adhesive molecules and integrins. According to the available data, MSCs are able to transmigrate through the endothelium and basement membrane into tissues [29]. It is interesting that the adhesion molecules CD106 (VCAM-1), CD54 (ICAM-1), and ICAM-2, which are found on endothelial cells, are also expressed by MSCs. Circulating MSCs are able to effectively travel to damaged sites through the adhesive molecules on their surface: CD24, CD29 (β1-integrin) and CD44. MSCs express the CD44 transmembrane glycoprotein, which can act as a ligand that mediates the adhesion of hyaluronic acid. The adhesion molecules late antigen-4 (VLA-4) and P-selectin are involved in MSC transmembrane migration. In addition, MSCs are characterized by the expression of multiple integrin receptors—α1, α2, α3, α4, α5, β1, β3 and β4—associated with cell–cell contacts and adhesion to extracellular matrix proteins. It is important to note the strong interaction of VLA-4/VCAM-1 particles, which are critical receptors for MSC transendothelial migration [31].
2.2.2. The Modulation of T Cells by MSCs
T-lymphocytes are among the main immune cells that influence inflammation in RA. Resident joint lymphocytes and MSCs that are present in the joint show mutual interactions. T lymphocytes directly affect MSCs, mainly through the effect of cytokines, IFN-γ and TNF-α, which stimulate the migration of MSCs, and at the same time block the differentiation capacity. MSCs have an inhibitory effect on the proliferation of T helper (Th) and cytotoxic T lymphocytes. In addition to the influence of cytokines, the subpopulation of Th1 and Th17 cells plays a significant role in degradation processes. IL-17-related cytokineTh17 suppresses the chondrogenic differentiation of MSCs by suppressing the chondrogenic transcription factor SRY-Box TF9 (SOX9) and its activator protein kinase A (PKA). The relationship between the influence of T cells and MSCs is reciprocal, and MSCs also influence T cells. MSCs have a stimulatory effect on the differentiation of Th2 and regulatory T cells (Treg), leading to an anti-inflammatory effect. In vitro culture MSCs have a suppressive effect on the proliferation of T lymphocytes in healthy individuals. MSCs have the effect of increasing the percentage of T-reg and act to maintain a tolerogenic immune state. Several conflicting results of the in vitro testing of RA patients’ MSCs to suppress T-lymphocyte proliferation suggest that the outcome could probably correlate with the disease severity [29].
2.2.3. Interactions between MSCs and Dendritic Cells
MSC have natural inhibitory effects on dendritic cells (DCs). They have a suppressive effect on the differentiation of monocytes into DCs. They also dampen the switch from monocytes to macrophages. This is related to the ability of MSCs to inhibit DC maturation and antigen presentation. MSCs expressing IL-1Rα can inhibit IL-1 production by DCs in co-cultures. It is a very interesting finding that RA DCs have a high expression of the pro-inflammatory transcription factor NF-κB, which is directly related to the production of inflammatory mediator TNF-α. It has been shown that monocyte-derived DCs can acquire a suppressor cell phenotype under the influence of MSCs. DC-modulated MSCs are capable of IL-10 and IL-4 secretion and the reduction of IL-12 and IFN-γ. DCs in the setting of RA indicate a failure of immune control mechanisms that could affect the activity of MSCs. MSCs could be involved in RA, and could act on DCs through TLR activation. TLR2 and TLR4 are activated in RA in the synovial environment. The cytokines IL-12 and IL-18 and IFN-γ act to increase TLR4. As a result, MSCs can produce pro-inflammatory IL-6. TLRs related to the pro-inflammatory functions of MSCs are evident in RA. MSCs and DCs behave differently in autoimmune diseases through pro-inflammatory cytokines [5][43].
References
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581.
- Trentham, D.E.; Dynesius-Trentham, R.A.; Orav, E.J.; Combitchi, D. Effects of oral administration of type II collagen on rheumatoid arthritis. Science 1993, 261, 1727–1730.
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361.
- Sarsenova, M.; Issabekova, A.; Abisheva, S.; Rutskaya-Moroshan, K.; Ogay, V. Mesenchymal Stem Cell-Based Therapy for Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 11592.
- De Bari, C. Are mesenchymal stem cells in rheumatoid arthritis the good or bad guys? Arthritis Res. Ther. 2015, 17, 9.
- van Delft, M.A.; Huizinga, T.W. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392.
- Holoshitz, J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr. Opin. Rheumatol. 2010, 22, 293.
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S. Meta-analysis: Diagnostic accuracy of anti–cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808.
- Van den Broek, M.; Dirven, L.; Klarenbeek, N.B.; Han, K.H.; Kerstens, P.J.S.M.; Huizinga, T.W.J. The association of treatment response and joint damage with ACPA-status in recent-onset RA: A subanalysis of the 8-year follow-up of the BeSt study. Ann. Rheum. Dis. 2012, 71, 245–248.
- van Boekel, M.A.; Vossenaar, E.R.; Van den Hoogen, F.H.; van Venrooij, W.J. Autoantibody systems in rheumatoid arthritis: Specificity, sensitivity and diagnostic value. Arthritis Res. Ther. 2001, 4, 7.
- Van Steendam, K.; Tilleman, K.; Deforce, D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology 2011, 50, 830–837.
- Musaelyan, A.; Lapin, S.; Nazarov, V.; Tkachenko, O.; Gilburd, B.; Mazing, A.; Mikhailova, L.; Shoenfeld, Y. Vimentin as antigenic target in autoimmunity. A comprehensive review. Autoimmun. Rev. 2018, 17, 926–934.
- Brentville, V.A.; Vankemmelbeke, M.; Metheringham, R.L.; Durrant, L.G. Post-translational modifications such as citrullination are excellent targets for cancer therapy. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2020; Volume 47, p. 101393.
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J. Exp. Med. 2009, 206, 449–462.
- Utz, P.J.; Genovese, M.C.; Robinson, W.H. Unlocking the “PAD” lock on rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 330–332.
- Zeltz, C.; Gullberg, D. Post-translational modifications of integrin ligands as pathogenic mechanisms in disease. Matrix Biol. 2014, 40, 5–9.
- Vossenaar, E.R.; Radstake, T.R.; van der Heijden, A.; van Mansum, M.A.; Dieteren, C. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381.
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069.
- Klareskog, L.; Rönnelid, J.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008, 26, 651–675.
- Demoruelle, M.K.; Deane, K. Antibodies to citrullinated protein antigens (ACPAs): Clinical and pathophysiologic significance. Curr. Rheumatol. Rep. 2011, 13, 421–430.
- Hill, J.A.; Bell, D.A.; Brintnell, W.; Yue, D. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J. Exp. Med. 2008, 205, 967–979.
- Luban, S.; Li, Z.G. Citrullinated peptide and its relevance to rheumatoid arthritis: An update. Int. J. Rheum. Dis. 2010, 13, 284–287.
- Machado, C.D.V.; Telles, P.D.D.S. Immunological characteristics of mesenchymal stem cells. Rev. Bras. De Hematol. E Hemoter. 2013, 35, 62–67.
- Friedenstein, A.J.; Latzinik, N.V.; Gorskaya, Y.F.; Luria, E.A.; Moskvina, I.L. Bone marrow stromal colony formation requires stimulation by haemopoietic cells. Bone Miner. 1992, 18, 199–213.
- Owen, M. Marrow stromal stem cells. J. Cell Sci. 1988, 10, 63–76.
- Caplan, A. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650.
- Procop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74.
- Reyes, M.; Lund, T.; Lenvik, T.; Aguiar, D.; Koodie, L.; Verfaillie, C.M. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood J. Am. Soc. Hematol. 2001, 98, 2615–2625.
- Si, Y.L.; Zhao, Y.L.; Hao, H.J.; Fu, X.B. MSCs: Biological characteristics, clinical applications and their outstanding concerns. Ageing Res. Rev. 2011, 10, 93–103.
- Azizi, S.A.; Stokes, D.; Augelli, B.J.; DiGirolamo, C. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats—similarities to astrocyte grafts. Proc. Natl. Acad. Sci. USA 1998, 95, 3908–3913.
- Scherzed, A.; Hackenberg, S. The differentiation of hMSCs counteracts their migration capability and pro-angiogenic effects in vitro. Oncol. Rep. 2016, 35, 219–226.
- Dominici, M.L.B.K.; Le Blanc, K.; Mueller, I. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317.
- Kuznetsov, S.A.; Mankani, M.H.; Bianco, P. Enumeration of the colony-forming units–fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. 2009, 2, 83–94.
- Mosna, F.; Sensebe, L.; Krampera, M. Human bone marrow and adipose tissue mesenchymal stem cells: A user’s guide. Stem Cells Dev. 2010, 19, 1449–1470.
- Parolini, O.; Alviano, F.; Bagnara, G.P. Concise review: Isolation and characterization of cells from human term placenta: Outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 2008, 26, 300–311.
- Soncini, M.; Vertua, E.; Gibelli, L.; Zorzi, F.; Denegri, M. Isolation and characterization of mesenchymal cells from human fetal membranes. J. Tissue Eng. Regen. Med. 2007, 1, 296–305.
- Bailo, M.; Soncini, M.; Vertua, E.; Signoroni, P.B. Engraftment potential of human amnion and chorion cells derived from term placenta. Transplantation 2004, 78, 1439–1448.
- Alviano, F.; Fossati, V.; Marchionni, C.; Arpinati, M. Term amniotic membrane is a high throughput source for multipotent mesenchymal stem cells with the ability to differentiate into endothelial cells in vitro. BMC Dev. Biol. 2007, 7, 1–14.
- Bačenková, D.; Rosocha, J.; Tóthová, T. Isolation and basic characterization of human term amnion and chorion mesenchymal stromal cells. Cytotherapy 2011, 13, 1047–1056.
- Shariatzadeh, S.; Shafiee, S.; Zafari, A.; Tayebi, T.; Yazdanpanah, G. Developing a pro-angiogenic placenta derived amniochorionic scaffold with two exposed basement membranes as substrates for cultivating endothelial cells. Sci. Rep. 2021, 11, 1–14.
- Jaramillo-Ferrada, P.A.; Wolvetang, E.J. Differential mesengenic potential and expression of stem cell-fate modulators in mesenchymal stromal cells from human-term placenta and bone marrow. J. Cell. Physiol. 2012, 227, 3234–3242.
- Koo, B.K.; Park, I.Y.; Kim, J.; Kim, J.H. Isolation and characterization of chorionic mesenchymal stromal cells from human full term placenta. J. Korean Med. Sci. 2012, 27, 857–863.
- Kurth, T.B.; Dell’Accio, F.; Crouch, V.; Augello, A.; Sharpe, P.T. Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Rheum. 2011, 63, 1289–1300.
- Lee, D.M.; Kiener, H.P.; Agarwal, S.K.; Noss, E.H.; Watts, G.F. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007, 315, 1006–1010.
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420.
More
Information
Subjects:
Rheumatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
843
Revisions:
2 times
(View History)
Update Date:
17 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No