 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Klaus Grossmann | -- | 5892 | 2022-08-10 14:21:49 | | | |
| 2 | Lindsay Dong | + 784 word(s) | 6676 | 2022-08-15 04:47:01 | | |
Video Upload Options
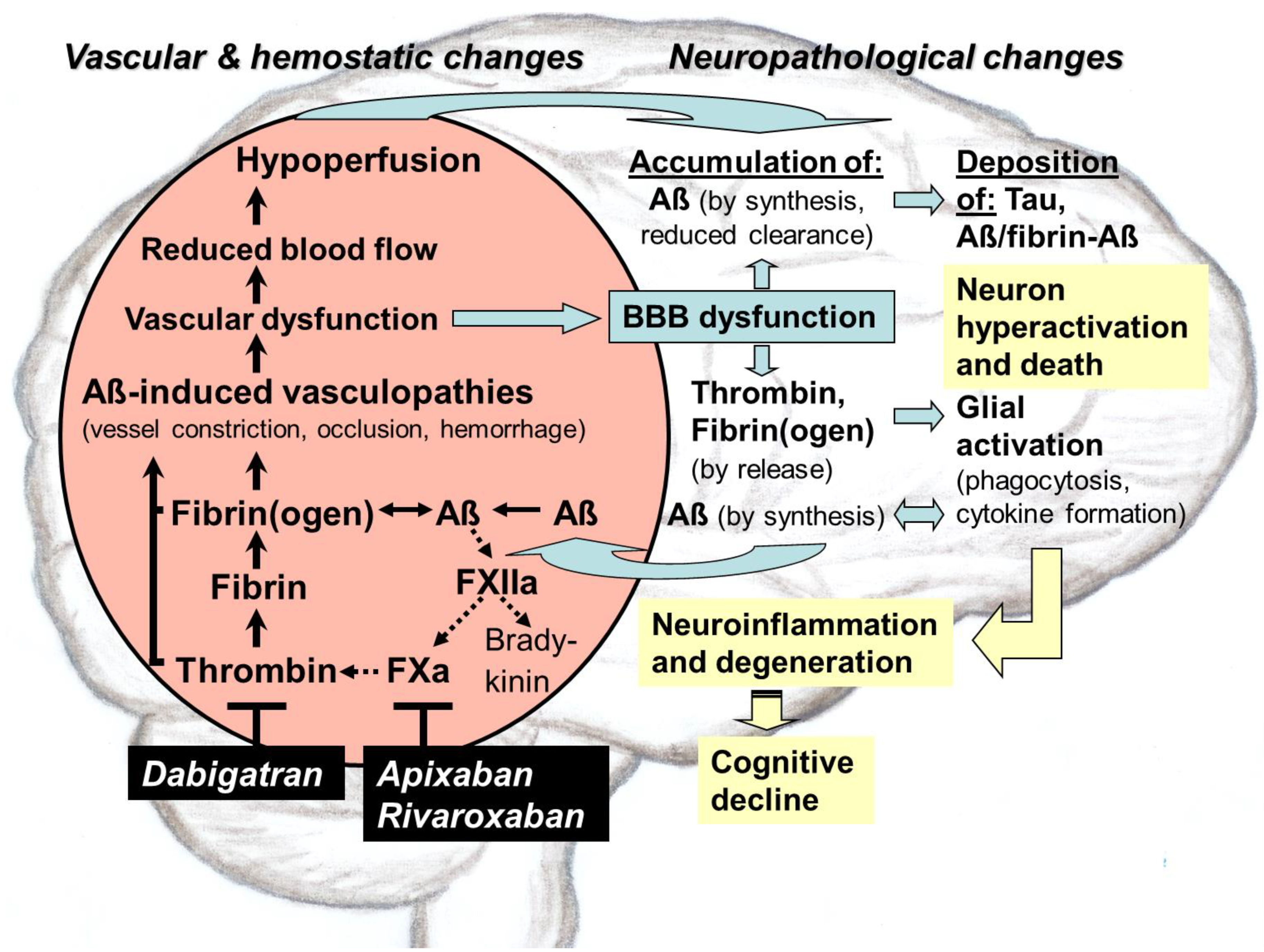
Direct oral anticoagulants (DOACs) target pathological thrombin, which is, like toxic tau and amyloid-ß proteins (Aß), an early hallmark of Alzheimer’s disease (AD). Especially in hippocampal and neocortical areas, the release of parenchymal Aß into the blood induces thrombin and proinflammatory bradykinin synthesis by activating factor XII of the contact system. Thrombin promotes platelet aggregation and catalyzes conversion of fibrinogen to fibrin, leading to degradation-resistant, Aß-containing fibrin clots. Together with oligomeric Aß, these clots trigger vessel constriction and cerebral amyloid angiopathy (CAA) with vessel occlusion and hemorrhages, leading to vascular and blood–brain barrier (BBB) dysfunction. As consequences, brain blood flow, perfusion, and supply with oxygen (hypoxia) and nutrients decrease. In parenchymal tissue, hypoxia stimulates Aß synthesis, leading to Aß accumulation, which is further enhanced by BBB-impaired perivascular Aß clearance. Aß trigger neuronal damage and promote tau pathologies. BBB dysfunction enables thrombin and fibrin(ogen) to migrate into parenchymal tissue and to activate glial cells. Inflammation and continued Aß production are the results. Synapses and neurons die, and cognitive abilities are lost. DOACs block thrombin by inhibiting its activity (dabigatran) or production (FXa-inhibitors, e.g., apixaban, rivaroxaban). Therefore, DOAC use could preserve vascular integrity and brain perfusion and, thereby, could counteract vascular-driven neuronal and cognitive decline in AD.
1. Introduction
2. Hemostasis, Thrombosis, and Antithrombotic Medication
2.1. Blood Coagulation and Fibrinolysis
2.2. Antithrombotic Therapy
2.2.1. Drug Portfolio
2.2.2. Fields of Indications
3. Toxic Proteins and Chronic Inflammation in AD
3.1. Generation and Occurrence of Aß
3.2. Brain Locations and Pathogenic Action of Aß
The preferential areas in human AD brain, where vascular Aß deposits and parenchymal Aß dense core plaques are diagnosed, are the neocortex and hippocampus, which are key for higher-order cognition, behavior, and motor skills [30][37]. The neocortex is involved in brain functions, which include sensory perception, motor commands, cognition and spatial reasoning, social and emotional behavior, memory, as well as learning and language processes. The hippocampus acts as a switchboard between perception and memory [38]. Early in AD, these cerebral areas show the dysfunction and hyperactivity of neurons, which are accompanied by progressing synapse and neuron cell death, closely correlated with the severity of cognitive impairment [28][39].
3.3. Tau Protein Pathologies
3.4. Inflammation and Glial Responses
4. Role of Aß in Triggering Vascular Constriction and CAA in AD
4.1. AD Mouse Models
4.2. Occurrence of CAA
4.3. Aß in CAA and Brain Parenchyma
4.4. Brain Vasculopathies and Lesions by Aß-Driven CAA
4.5. Pathophysiological Impact of Aß on Vascular and BBB Functioning
5. Interaction of Aß with the Plasma Contact System and its Driven Pathways of Coagulation and Inflammation in AD
5.1. Aß-Induced Activation of FXII in Contact System and Effects on Pathways Beyond
5.2. Pathological Dimension
6. Therapeutical Intervention Using Thrombin-Inhibiting Anticoagulants against Dysregulated Intrinsic Coagulation in AD
The first clinical studies on anticoagulant medication against dementia in small groups of senile-presenile patients extend far back into the 1960s and resulted in decelerated cognitive decline and reduced morbidity and mortality [73][74][75][76]. In addition, over the last 20 years, multiple observer studies on patients with anticoagulant use due to atrial fibrillation (AF) suggest that oral anticoagulants, particularly DOACs, safeguard against incidence of dementia like AD, as shown in persons without a dementia history before treatment [77][78][79][80][81][82][83][84].
7. Clinical Perspective for Anticoagulant Use against AD
7.1. Evaluation of Therapeutic Suitability of Available Anticoagulants
The parenteral administration of a drug by injection or infusion directly into the blood stream are less suitable for a permanent therapy from their handling alone. This would be the case when treating parenteral, indirect thrombin-inhibiting heparins (e.g., enoxaparin), heparinoid danaparoid sodium, and fondaparinux or direct thrombin-inhibiting hirudin, bivalirudin, and the synthetic L-arginine derivative argatroban. Heparins, hirudin, and derivatives are usually administered for short-term prophylaxis of thromboembolic events and for therapy of acute venous thrombosis [16]. Undesirable side effects of treatment with heparins are given by abnormally low levels of platelets in the blood (thrombocytopenia) and increased risk of bleeding. In addition, anticoagulation by heparins is unpredictably affected by unspecific plasma protein binding. Furthermore, heparins do not inhibit fibrin-bound thrombin and related thrombus formation [16][85]. In contrast to heparins, hirudin inactivates thrombin bound to clots and does not directly interact with platelets. However, in therapeutic application, bleeding complications have been frequently observed [86].
With respect of administration and handling, oral treatment with anticoagulants for therapy would certainly be preferable to a parenteral application, if the efficacy and safety profile of the oral drug is also favorable and the patient can take the medication on a consistent basis. For oral administration, anticoagulants are available on the market from VKA-type, such as warfarin, phenprocoumon, as well as from DOAC-type with the direct thrombin inhibitor dabigatran and the direct FXa inhibitors apixaban, betrixaban, edoxaban, rivaroxaban [16]. Dabigatran, rivaroxaban, and apixaban were approved for antithrombotic use already at the beginning of the 2010s years [16]. Approval of edoxaban and, subsequently, betrixaban was in the second half of the decade [16]. The development of the new class of antithrombotic DOACs that directly target to specific factors in intrinsic coagulation was particularly desired due to serious disadvantages of the conventional VKAs [16][19][87].
Compared to VKAs, DOACs provide constant therapeutic efficacy and a more favorable safety profile, as well as avoidance of adverse effects from vitamin K deficiency due to their different mechanism of action. In detail, the advantages of DOACs include (i) rapid onset of action, (ii) short half-life, (iii) less drug–drug interactions and no dietary interactions, and (iv) safe antidote strategies in situations of bleeding risk. Therefore, the lower intra- and interindividual variability in the DOAC-effect allows fixed dosing and a predictable anticoagulative response without the need for continuous monitoring of the drug level in patients [9][11][16][19][87][88][89]. Since DOACs, especially dabigatran, are eliminated to a large extent via the kidney, the renal function in patients should be routinely monitored, particularly in elderly persons due to increasing renal impairment and associated co-morbidities [19][87]. In patients with renal impairment, dependent on severity, DOAC use requires dose adjustments or is contraindicated [19][87]. In addition, DOAC-type anticoagulants also hold the risk of bleeding, particularly of serious intracranial hemorrhage [16][19][87].
In a systematic review and meta-analysis of phase III trials for stroke and systemic embolism prevention in patients with AF (2009–2013), DOACs (apixaban, dabigatran, edoxaban, rivaroxaban) showed a more favorable risk–benefit profile compared with warfarin [90]. In addition, reductions in all-cause mortality and systemic embolic events, these agents reduced the risk of hemorrhagic stroke by 51% and the risk of intracranial hemorrhage by 52% [90]. This favorable efficacy and safety profile was consistent with many subgroups and ethnicities including the Asian population [87][90]. Conversely, dose-dependently, the risk of gastrointestinal bleeding was 25% higher with DOACs than with warfarin [90]. Similar results were obtained in a large retrospective observational study of ca. 400,000 AF patients, based on US claims data (2013–2015) [89]. Treatment with DOACs (dabigatran, apixaban, rivaroxaban) was associated with lower rates of stroke and systemic embolism, compared with warfarin. In addition, apixaban and dabigatran showed lower rates of major bleeding, including gastrointestinal bleeding, intracranial hemorrhage, and major bleeding at other key sites, whereas rivaroxaban had a higher rate of major bleeding, compared with warfarin [89]. This is in accordance with results from a new-user retrospective cohort study of patients with AF and dementia (2011–2017), comparing DOAC treatment versus warfarin [91]. DOAC-treated patients, who were older and had more comorbidities than the warfarin-treated individuals, showed similar prevention of thromboembolic events, compared to warfarin, but a reduced risk of intracranial bleeding [91]. However, the risk of gastrointestinal bleeding was increased in the DOAC treatment [91].
7.2. DOAC-Type Anticoagulants for In-Depth Clinical Investigation
Altogether, the wide range of clinical observer studies, conducted in the last 15 years particularly in elderly individuals with AF, demonstrate that DOAC-type anticoagulants exhibit a predictable therapeutic effect in preventing stroke and systemic embolic events. In addition, DOACs show a safety profile that more than halves the risk of dangerous intracranial hemorrhage in elderly people compared to VKAs [87][90][92]. These properties, along with their pharmacological advantages, give DOACs a clear preference over VKAs, particularly when anticoagulants are administered to elderly people, who are more vulnerable individuals. In addition, gastrointestinal bleedings, which are more likely to be promoted by DOACs compared to VKAs [87][90][91], can be treated better and stopped immediately after occurrence by effective antidote strategies. These strategies have been successfully developed for various DOACs in the recent years [16]. In the case of dabigatran, the specific antidote idarucizumab (Praxbind®) was introduced in 2016 [93]. This antibody binds to dabigatran with high affinity and leads within minutes to a rapid cancellation of the anticoagulative effect, e.g., in emergency operations or in situations of uncontrollable bleeding [16][93]. In the case of the FXa inhibitors apixaban and rivaroxaban, andexanet alfa (AndexXa®) was recently approved as a fast-acting antidote [16][94]. Andexanet alfa is a recombinantly modified, human FXa molecule, which itself has no effect on blood clotting. It acts as a kind of decoy protein that binds the FXa-inhibitors and thus restores blood clotting [16][94]. Generally, availability of an efficient and fast acting reversal agent should be a prerequisite for a long-term anticoagulative treatment. This is especially the case in elderly and comorbid AD patients, showing inherent bleeding risk due to fragile blood vessels [57].
References
- Bickel, H. Die Häufigkeit von Demenzerkrankungen. Inf. Dtsch. Ges. Selbsthilfe Demenz Berl. 2020, 1, 1–10.
- Abbott, A. Treating Alzheimer’s before it takes hold. Nature 2022, 603, 216–219.
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66.
- Jeremic, D.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Past, present and future of therapeutic strategies against amyloid-ß peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496.
- Strickland, S. Blood will out: Vascular contributions to Alzheimer’s disease. J. Clin. Investig. 2018, 128, 556–563.
- Zhu, C.; Chen, Y.; Mao, R.; Wang, J.; Liu, R.; Liu, Y.; Wang, X. Advances in drug therapy for Alzheimer’s disease. Curr. Med. Sci. 2020, 40, 999–1008.
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310.
- Bauzon, J.; Lee, G.; Cummings, J. Repurposed agents in the Alzheimer’s disease drug development pipeline. Alzheimer’s Res. Ther. 2020, 13, 98.
- Grossmann, K. Anticoagulants for treatment of Alzheimer’s disease. J. Alzheimers Dis. 2020, 77, 1373–1382.
- Grossmann, K. Direct oral anticoagulants: A new therapy against Alzheimer’s disease? Neural. Reg. Res. 2021, 16, 1556–1557.
- Grossmann, K. Alzheimer’s disease—Rationales for potential treatment with the thrombin inhibitor dabigatran. Int. J. Mol. Sci. 2021, 22, 4805.
- Scheffer, S.; Hermkens, A.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular hypothesis of Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283.
- Singh, P.K.; Badimon, A.; Chen, Z.-L.; Strickland, S.; Norris, E.H. The contact activation system and vascular factors as alternative targets for Alzheimer’s disease therapy. Res. Pract. Thromb. Haemost. 2021, 5, e12504.
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892.
- Grossmann, K. Alzheimer-Krankheit–können Antikoagulantien helfen? J. Neurol. Neurochir. Psychiat. 2021, 22, 7–10.
- Grosser, T.; Weber, A.-A. Pharmakologie der Hämostase. In Allgemeine und spezielle Pharmakologie und Toxikologie, 12th ed.; Aktories, K., Förstermann, U., Hofmann, F., Starke, K., Eds.; Elsevier: München, Germany, 2017; pp. 465–488.
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. 2018, 19, 283–301.
- Martin, R. Targeting fibrin in neurodegeneration. Nat. Immunol. 2018, 19, 1149–1150.
- Shameem, R.; Ansell, J. Disadvantages of VKA and requirements for novel anticoagulants. Best. Pract. Res. Clin. Haematol. 2013, 26, 103–114.
- Klimke, K.; Paschke, L.; Schulz, M. Orale Antikoagulantien. In Rx-Trendbericht: Thema im Fokus; Zentralinstitut für die Kassenärztliche Versorgung in Deutschland: Berlin, Germany, 2019; pp. 1–5.
- Kresge, N.; Simoni, R.D.; Hill, R.L. Hemorrhagic sweet clover disease, dicumarol, and warfarin: The work of Karl Paul Link. J. Biolog. Chem. 2005, 280, e5–e6.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135.
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498.
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51.
- Goedert, M. Alzheimer’s and parkinson’s diseases: The prion concept in relation to assembled Aß, tau, and α-synuclein. Science 2015, 349, 601.
- Yamada, M. Cerebral amyloid angiopathy: Emerging concepts. J. Stroke 2015, 17, 17–30.
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The physiological roles of amyloid-ß peptide hint at new ways to treat Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 118.
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.-J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of ß amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565.
- Selkoe, D.J. Treatments for Alzheimer’s disease emerge. Science 2021, 373, 624–626.
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-ß 42 filaments from human brain. Science 2022, 375, 167–172.
- Lattanzi, V.; Andre, I.; Gasser, U.; Dubackic, M.; Olsson, U.; Linse, S. Amyloid-ß 42 fibril structure based on small-angle scattering. Proc. Natl. Acad. Sci. USA 2021, 118, e2112783118.
- Ghosh, U.; Yau, W.-M.; Collinge, J.; Tycko, R. Structural differences in amyloid-ß fibrils from brains of nondemented elderly individuals and Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2111863118.
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191.
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s disease and vascular aging. J. Am. Coll. Cardiol. 2020, 75, 942–951.
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167.
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42.
- Huffman, K. The developing, aging neocortex: How genetics and epigenetics influence early developmental patterning and age-related change. Front. Genet. 2012, 3, 212.
- Treder, M.S.; Chares, I.; Michelmann, S.; Martin-Buro, M.C.; Roux, F.; Carceller-Benito, F.; Ugalde-Canitrot, A.; Rollings, D.T.; Sawlani, V.; Chelvarajah, R.; et al. The hippocampus as the switchboard between perception and memory. Proc. Natl. Acad. Sci. USA 2021, 118, e2114171118.
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hipocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384.
- Chang, C.-W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255.
- He, Z.; Guo, J.L.; McBridge, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-ß plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38.
- Lee, M.-S.; Tsai, L.-H. Cdk5: One of the links between senile plaques and neurofibrollary tangles? J. Alzheimers Dis. 2003, 5, 127–137.
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropath. 2020, 140, 793–810.
- Brown, G.C.; St George-Hyslop, P.H. Deciphering miocroglial diversity in Alzheimer’s disease. Science 2017, 356, 1123–1124.
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellivol, M.; Brown, G.C. Microglia phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639.
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia- derived ASC specks cross-seed amyloid-ß in Alzheimer’s disease. Nature 2017, 552, 355–361.
- Hur, J.-Y.; Frost, G.F.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740.
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154.
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 2021, 595, 701–706.
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katz-Marski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204.
- Schoch, K.M.; Ezerskiy, L.A.; Morhaus, M.M.; Bannon, R.N.; Sauerbeck, A.D.; Shabsovich, M.; Jafar-nejad, P.; Rigo, F.; Miller, T.M. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2100356118.
- Profaci, C.P.; Munij, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150.
- Broce, I.J.; Tan, C.H.; Fan, C.C.; Jansen, I.; Savage, J.E.; Witoelar, A.; Wen, N.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropath. 2019, 137, 209–226.
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12.
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural. Transm. 2002, 109, 813–836.
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R., Jr. Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182.
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoli, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-ß pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250.
- Sweeney, M.D.; Zlokovic, B.V. Lymphatic waste disposal in the brain. Nature 2018, 560, 172–174.
- Li, H.; Guo, Q.; Inoue, T.; Polito, V.A.; Tabuchi, K.; Hammer, R.E.; Pautler, R.G.; Taffet, G.E.; Zheng, H. Vascular and parenchymal amyloid pathology in an Alzheimer’s disease knock-in mouse model: Interplay with cerebral blood flow. Mol. Neurodegener. 2014, 9, 28.
- Maier, F.C.; Wehrl, H.F.; Schmid, A.M.; Mannheim, J.G.; Wiehr, S.; Lerdkrait, C.; Calaminus, C.; Stahlschmidt, A.; Ye, L.; Burnet, M.; et al. Longitudinal PET-MRI reveals ß-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med. 2014, 20, 1485–1492.
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Perez, J.M.; Evans, A.C. The Alzheimer’s Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934.
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid ß oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518.
- Wolters, F.J.; Zonneveld, H.I.; Hofman, A.; van der Lugt, A.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Cerebral perfusion and the risk of dementia. Circulation 2017, 136, 719–728.
- Roher, A.E.; Debbins, J.P.; Malek-Ahmadi, M.; Chen, K.; Pipe, J.G.; Maze, S.; Belden, C.; Maarouf, C.L.; Thiyyagura, P.; Mo, H.; et al. Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 2012, 8, 599–611.
- Singh, P.K.; Chen, Z.-L.; Strickland, S.; Norris, E.H. Increased contact system activation in mild cognitive impairment patients with impaired short-term memory. J. Alzheimers Dis. 2020, 77, 59–65.
- Zamolodchikov, D.; Renne, T.; Strickland, S. The Alzheimer’s disease peptide ß-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007.
- Zamolodchikov, D.; Strickland, S. A possible new role for Aß in vascular and inflammatory dysfunction in Alzheimer’s disease. Thromb. Res. 2016, 141 (Suppl. S2), S59–S61.
- Zamolodchikov, D.; Chen, Z.-L.; Conti, B.A.; Renne, T.; Strickland, S. Activation of the factor XII-driven contact system in Alzheimer’s disease patient and mouse model plasma. Proc. Natl. Acad. Sci. USA 2015, 112, 4068–4073.
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: Implications for Alzheimer’s disease. Front. Neurosci. 2020, 14, 762.
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nueda, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.; et al. Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J. Am. Coll. Cardiol. 2019, 74, 1910–1923.
- Khalil, R.B. Direct thrombin inhibitor’s potential efficacy in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2012, 27, 564–567.
- Whittier, J.R.; Korenyi, C.; Klein, D.F.; Foley, W. Prevention of degenerative disease: A controlled study of anticoagulant prophylaxis. J. Chronic Dis. 1961, 14, 203–212.
- Ratner, J.; Rosenberg, G.; Kral, V.A.; Engelsmann, F. Anticoagulant therapy for senile dementia. J. Am. Geriatr. Soc. 1972, 20, 556–559.
- Walsh, A.C.; Walsh, B.H.; Melaney, C. Senile-presenile dementia: Follow-up data on an effective psychotherapy-anticoagulant regimen. J. Am. Geriatr. Soc. 1978, 26, 467–470.
- Barber, M.; Tait, C.; Scott, J.; Rumley, A.; Lowe, D.O.; Stott, D.J. Dementia in subjects with atrial fibrillation: Hemostatic function and the role of anticoagulation. J. Throm. Haemost 2004, 2, 1873–1878.
- Friberg, L.; Rosenqvist, M. Less dementia with oral anticoagulation in atrial fibrillation. Eur. Heart J. 2018, 39, 453–460.
- Mongkhon, P.; Fanning, L.; Lau, W.C.Y.; Tse, G.; Lau, K.K.; Wei, L.; Kongkaew, C.; Wong, I.C.K. Oral anticoagulant and reduced risk of dementia in patients with atrial fibrillation: A population-based cohort study. Heart Rhythm 2020, 17, 706–713.
- Jacobs, V.; May, H.T.; Bair, T.L.; Crandall, B.G.; Cutler, M.J.; Day, J.D.; Mallender, C.; Osborn, J.S.; Stevens, S.M.; Weiss, J.P.; et al. Long-term population-based cerebral ischemic event and cognitive outcomes of direct oral anticoagulants compared with warfarin among long-term anticoagulated patients for atrial fibrillation. Am. J. Cardiol. 2016, 118, 210–214.
- Cadogan, S.L.; Powell, E.; Wing, K.; Wong, A.Y.; Smeeth, L.; Warren-Gash, C. Anticoagulant prescribing for atrial fibrillation and risk of incident dementia. Heart 2021, 107, 1898–1904.
- Cheng, W.; Liu, W.; Li, B.; Li, D. Relationship of anticoagulant therapy with cognitive impairment among patients with atrial fibrillation. A meta-analysis and systemic review. J. Cardiovasc. Pharmocol. 2018, 71, 380–387.
- Zeng, D.; Jiang, C.; Su, C.; Tan, Y.; Wu, J. Anticoagulation in atrial fibrillation and cognitive decline. Medicine 2019, 98, e14499.
- Mongkhon, P.; Naser, A.Y.; Fanning, L.; Tse, G.; Lau, W.C.Y.; Wong, I.C.K.; Kongkaew, C. Oral anticoagulants and risk of dementia: A systematic review and meta-analysis of observational studies and randomized controlled trials. Neurosci. Biobehav. Rev. 2019, 96, 1–9.
- Ho, B.-L.; Hsieh, S.-W.; Chou, P.-S.; Yang, Y.-H. Effects of dabigatran on dementia pathogenesis and neuropsychological function: A review. J. Alzheimers Dis. 2022, 86, 1589–1601.
- Bates, S.M.; Weitz, J.I. The mechanism of action of thrombin inhibitors. J. Invasive Cardiol. 2000, 12, F27–F32.
- Monreal, M.; Costa, J.; Salva, P. Pharmacological properties of hirudin and its derivatives. Potential clinical advantages over heparin. Drugs Aging 1996, 8, 171–182.
- Zirlik, A.; Bode, C. Vitamin K antagonists: Relative strengths and weaknesses vs. direct oral anticoagulants for stroke prevention in patients with atrial fibrillation. J. Thromb. Thrombolysis 2017, 43, 365–379.
- Van Ryn, J.; Goss, A.; Hauel, N.; Wienen, W.; Priepke, H.; Nar, H.; Clemens, A. The discovery of Dabigatran etexilate. Front. Pharmacol. 2013, 4, 12.
- Lip, G.Y.H.; Keshishian, A.; Li, X.; Hamilton, M.; Masseria, C.; Gupta, K.; Luo, X.; Mardekian, J.; Friend, K.; Nadkarni, A.; et al. Effectiveness and safety of oral anticoagulants among nonvalvular atrial fibrillation patients. The ARISTOPHANES Study. Stroke 2018, 49, 2933–2944.
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962.
- Fanning, L.; Lau, W.C.Y.; Mongkhon, P.; Man, K.K.C.; Bell, J.S.; Ilomäki, J.; Darzins, P.; Lau, K.K.; Wei, L.; Wong, I.C.K. Safety and effectiveness of direct oral anticoagulants vs warfarin in people with atrial fibrillation and dementia. J. Am. Med. Dir. Assoc. 2020, 21, 1058–1064.
- Graham, D.J.; Reichman, M.E.; Wernecke, M.; Zhang, R.; Southworth, M.R.; Levenson, M.; Sheu, T.-C.; Mott, K.; Goulding, M.R.; Houstoun, M.; et al. Cardiovascular, bleeding, and mortality risks in elderly medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation 2015, 131, 157–164.
- Pollack, C.V., Jr.; Paul, M.D.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Hulsman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for dabigatran reversal. N. Engl. J. Med. 2015, 373, 511–520.
- Connolly, S.J.; Truman, J.M., Jr.; Eikelboom, J.W.; Gibson, C.M.; Curnutte, J.T.; Gold, A.; Bronson, M.D.; Lu, G.; Conley, P.B.; Verhamme, P.; et al. for the ANNEXA-4 investigators Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N. Engl. J. Med. 2016, 375, 1131–1141.

