Direct oral anticoagulants (DOACs) target pathological thrombin, which is, like toxic tau and amyloid-ß proteins (Aß), an early hallmark of Alzheimer’s disease (AD)AD. Especially in hippocampal and neocortical areas, the release of parenchymal Aß into the blood induces thrombin and proinflammatory bradykinin synthesis by activating factor XII of the contact system. Thrombin promotes platelet aggregation and catalyzes conversion of fibrinogen to fibrin, leading to degradation-resistant, Aß-containing fibrin clots. Together with oligomeric Aß, these clots trigger vessel constriction and cerebral amyloid angiopathy (CAA) with vessel occlusion and hemorrhages, leading to vascular and blood–brain barrier (BBB) dysfunction. As consequences, brain blood flow, perfusion, and supply with oxygen (hypoxia) and nutrients decrease. In parenchymal tissue, hypoxia stimulates Aß synthesis, leading to Aß accumulation, which is further enhanced by BBB-impaired perivascular Aß clearance. Aß trigger neuronal damage and promote tau pathologies. BBB dysfunction enables thrombin and fibrin(ogen) to migrate into parenchymal tissue and to activate glial cells. Inflammation and continued Aß production are the results. Synapses and neurons die, and cognitive abilities are lost. DOACs block thrombin by inhibiting its activity (dabigatran) or production (FXa-inhibitors, e.g., apixaban, rivaroxaban). Therefore, DOAC use could preserve vascular integrity and brain perfusion and, thereby, could counteract vascular-driven neuronal and cognitive decline in AD.
- Alzheimer´s disease
- blood–brain barrier dysfunction
- inflammation
- vascular dysfunction
- tau
- thrombin
- fibrin
- direct oral anticoagulants
1. Introduction
2. Hemostasis, Thrombosis, and Antithrombotic Medication
2.1. Blood Coagulation and Fibrinolysis
In hemostasis, the vascular wall, platelets, and the coagulation and fibrinolysis system in the blood interact functionally together in order to (i) quickly seal injured vessels with a clot of blood, a thrombus, and thus stopping bleeding; (ii) limit thrombus formation to the area of vascular injury; and (iii) remove the thrombus during wound healing [16]. The formation of a thrombus involves platelet activation through endothelial cells of the damaged blood vessel and the initiation of multiple, closely linked and regulated biochemical processes in the plasmatic coagulation system via extrinsic and intrinsic pathways [16]. At the common connection of both pathways, activation of the key enzyme of blood clotting, the serine protease thrombin (factor IIa), takes place by the prothrombinase complex. Thrombin cleaves fibrinogen, a soluble protein, which then polymerizes to insoluble fibrin protofibrils. Fibrinogen normally circulates in the blood in huge quantities, released particularly by the liver. Fibrin protofibrils form a stable construct of cross-linked fibrin strands with integrated erythrocytes and platelets, known as a blood clot or thrombus. In addition, fibrin(ogen) can trigger inflammatory processes in neurodegeneration [17][18][17,18].2.2. Antithrombotic Therapy
2.2.1. Drug Portfolio
Overall, thrombotic events are a major complication of cardiovascular diseases, with fatal consequences if not appropriately treated by antithrombotic medication [16]. The available portfolio comprises three categories of drugs: 1. platelet aggregation inhibitors (PAIs), which prevent blood clots in long-term management of arterial thrombosis by blocking cyclooxygenase (COX) or a platelet surface receptor; 2. fibrinolytics, which permit lysis of an already formed thrombus and thereby vessel reperfusion by converting plasminogen to fibrin-dissolving plasmin; and 3. anticoagulants [16]. Nowadays, a palette of anticoagulants with different dosages, forms, and mechanisms of action is available, which can affect the plasmatic coagulation cascade indirectly, as in the case of the long-known, oral-active VKAs and of parenteral heparins (e.g., enoxaparin, a low molecular weight glycosaminoglycan form of heparin), heparinoid danaparoid sodium, and fondaparinux [16]. Heparins inactivate thrombin and FXa through an antithrombin-dependent mechanism [16]. On the other hand, both components of the coagulation cascade can also be inhibited directly, independent of antithrombin. Thus, thrombin is directly blocked, e.g., by parenteral hirudin, bivalirudin, argatroban. Likewise, orally active dabigatran binds to and thus directly inhibits both soluble and fibrin-bound thrombin [16]. Dabigatran is administered as the prodrug form dabigatran etexilate (Pradaxa®). On the other hand, for direct FXa inhibition, oral-active drugs, including apixaban (Eliquis®), betrixaban (Bevyxxa®), edoxaban (Lixiana®), and rivaroxaban (Xarelto®), are available. They bind, selectively and reversibly, to the active site of activated factor FX (FXa) in an antithrombin-independent manner [16]. Both the direct thrombin-inhibitor dabigatran and the FXa-inhibitors represent the newest category of anticoagulants, summarized as direct oral anticoagulants (DOACs). These specific-acting DOACs, which were launched into the pharmaceutical market in the last 15 years, are characterized by easy dosing regiments, no food–drug interactions, and no need for constant patient monitoring, as well as by a reduced risk of dangerous intracranial bleeding [9][11][16][19][9,11,16,20]. DOACs are approved for a broad spectrum of cardiovascular indications, in order to prevent stroke and systemic embolism in patients, who exhibit, e.g., nonvalvular atrial fibrillation (AF) or increased cardiovascular risk factors, such as heart failure and high blood pressure. Likewise, DOAC therapy and prophylaxis in deep vein thrombosis and pulmonary embolism is indicated [16][19][16,20]. Today, DOACs are prescribed for antithrombotic treatments alone in Germany in approximately 2 million patients, mostly over 70 years of age [16][20][16,21]. In contrast, the number of prescriptions of VKA-type anticoagulants decreased in the last years and currently amounts to approximately 1 million patients in Germany [20][21].2.2.2. Fields of Indications
Currently, anticoagulants are used as short- and long-term options to prevent and to treat blood clots that may occur in blood vessels and cause stroke and thromboembolism events [16]. Short-term treatment is required in acute venous thrombosis but also for prophylaxis of thrombotic events in risk situations, such as surgeries. Long-term to permanent anticoagulation is ordained for the prevention of thromboembolic complications in patients who show heart arrhythmias (e.g., AF) and cardiovascular and thrombophilic risk factors or are equipped with a mechanical heart valve substitute [16]. Particularly, patients with AF have a five-fold increased incidence of stroke, which is associated with a high risk of disability and death [16][21][16,19]. The benefit of the antithrombotic prophylaxis and therapy is high and drastically reduces the risk of myocardial infarction, ischemic stroke, and mortality in vulnerable persons. Nevertheless, a certain risk of intracranial and gastrointestinal hemorrhage must be taken into account when treatment is prescribed [16]. Recently, a completely new aspect for application of anticoagulants has come into focus, which is based on new findings about the impact of vascular and hemostatic dysfunction in AD pathogenesis. Therefore, medical repositioning of anticoagulants towards therapeutic and prophylactic use for AD is in discussion, and clinical studies for approval are recommended [8][9][10][11][15][8,9,10,11,15].3. Toxic Proteins and Chronic Inflammation in AD
3.1. Generation and Occurrence of Aß
The accumulation of misfolded, toxic oligomers of Aß in the AD brain is thought to be the result of pathological dyshomeostasis between progressive Aß production and failure of their clearance. In the “amyloid hypothesis of AD”, Aß are seen in the center of the key factors that initiate disease pathogenesis with its cascade of events, which include the development of tau neurofibrillary tangles, oxidative stress, and inflammatory and neurodegenerative processes [22][23][24][25][26][27][28][29][30][23,24,25,26,27,28,29,30,31]. Currently, this hypothesis provides the most important starting point for the search of novel drugs that can possibly slow, stop, cure, or prevent the disease [4][23][27][29][4,24,28,30]. In the amyloidogenic pathway for the generation of toxic Aß, Aß are excised from a single-domain membrane protein, the amyloid-ß precursor protein (AßPP), which is cleaved by sequentially acting transmembrane proteases, the ß- and γ-secretase [22][23][24][25][26][27][28][29][30][23,24,25,26,27,28,29,30,31]. Particularly, mutations in the Aß region of the AßPP precursor gene and in the presenilin subunits of the γ-secretase lead to aggressive Aß forms. These toxic Aß species are associated with early onset familial AD and vary in size, including Aß isoforms of 40 (Aß40) and 42 (Aß42) amino acids as the most abundant. A third AßPP splitting enzyme, the α-secretase, is involved in a nonamyloidogenic pathway, which does not contribute to the production of amyloid plaques. AßPP is mainly embedded in the plasma membrane of different types of neurons and glial cells. After secretase cleavage of AßPP, extracellular, soluble fragments of different length are produced, which include secreted AßPP fragment and Aß isoforms. Among these isoforms, oligomeric Aß40 of the shorter subtype is the predominant one, while Aß42 is more neurotoxic and aggregates faster than Aß40. Aß42 is the major species observed in plaques. Aß42 oligomers are also able to enter cells via endocytosis and to cause lysosomal fusion dysfunction. This effect can lead to increasing excretion of modified Aß into the extracellular space and can reduce Aß elimination by microglia phagocytosis [23][24][25][26][27][28][29][30][24,25,26,27,28,29,30,31]. At physiological level in the healthy brain, soluble Aß (predominantly Aß40, Aß42) and AßPP fragments are thought to be required for (i) synaptic functioning and neuronal survival [4]; (ii) repairing leaks in the vascular interface to the brain, the blood–brain barrier (BBB); and (iii) for the defense against pathogen infections [27][28]. In conditions of excessive Aß generation in the diseased AD brain, toxic Aß are secreted into the extracellular space, and accumulate initially as self-aggregating Aß monomers into soluble dimers, fibrillar oligomers, and polymers (protofibrils). Then, they are deposited as insoluble fibrils and fibrillar Aß plaques between neurons. Especially, fibrillar Aß seem to be a reservoir and source of toxicity to neuronal cells because the fibril surface can catalyze the conversion of Aß monomers into toxic oligomeric species [31][32]. In addition, multiple distinct fibril structures, called fibril polymorphs, are generated by the different Aß isoforms, which may differ in their neurotoxic potential [32][33]. Parenchymal Aß are transported within the brain´s interstitial fluid (ISF) along the walls of blood vessels to the meningeal cerebrospinal fluid (CSF) and lymphatic vessels. The removal of parenchymal Aß is primarily from the ISF by transfer into the blood vessels across the BBB [33][34]. This process is called perivascular Aß clearance [33][34]. With increasing parenchymal Aß release into the blood, Aß oligomers (in particular subtype Aß40) deposit around and in the walls of leptomeningeal and cortical blood vessels. This cerebrovascular Aß cause a disease known as Aß-type cerebral amyloid angiopathy (CAA), which is associated with AD pathogenesis [5][26][33][34][35][36][5,27,34,35,36,37]. CAA affects vascular activity (vasoactivity) and functioning, which again also interferes with perivascular Aß clearance. Concomitantly, vascular Aß clearance is additionally impaired by a decrease in the diameter of meningeal lymphatic vessels for Aß drainage. This further amplifies Aß accumulation in the brain parenchyma.3.2. Brain Locations and Pathogenic Action of Aß
The preferential areas in human AD brain, where vascular Aß deposits and parenchymal Aß dense core plaques are diagnosed, are the neocortex and hippocampus, which are key for higher-order cognition, behavior, and motor skills [30][37][31,38]. The neocortex is involved in brain functions, which include sensory perception, motor commands, cognition and spatial reasoning, social and emotional behavior, memory, as well as learning and language processes. The hippocampus acts as a switchboard between perception and memory [38][39]. Early in AD, these cerebral areas show the dysfunction and hyperactivity of neurons, which are accompanied by progressing synapse and neuron cell death, closely correlated with the severity of cognitive impairment [28][39][29,40].
3.3. Tau Protein Pathologies
The accumulation of Aß in the AD brain precedes intraneural deposition of tau proteins, which has been found to also be a protein hallmark of illness. Toxic aggregates of this protein, which spread via neuron-to-neuron connections throughout the brain, are also typically observed in connection with neurodegenerative events in AD [29][40][30,45]. In the diseased brain, axonal, microtubule-associated tau proteins are increasingly phosphorylated by kinases. Hyperphosphorylated tau is able to aggregate to insoluble, filamentous tau structures, which form neurofibrillary tangles (NFTs) in the brain. The hyperphosphorylation of tau is increased by the accumulation of oligomeric Aß, which activate (i) kinases for tau hyperphosphorylation and (ii) inactivate phosphatases for tau dephosphorylation. The hyperphosphorylation of tau leads to tau relocation from axonal microtubules to dendrites [4][41][42][4,46,47]. Tau tangles, fragments, and oligomeric aggregates accumulate in neuronal bodies and synapses, where tau interferes with glutamate receptor trafficking and associated excitation, as well as with neuronal firing. The disruption of synaptic function, accompanied by Aß- and tau-induced loss of axonal myelin, are followed by loss of synapses and neurons. Thereby, neuron cell death is based on apoptosis and neurotransmitter deficiency, and precedes cognitive decline. The hyperphosphorylation of tau has also been related to the decrease in cerebral blood flow (CBF), a further hallmark of AD [40][43][45,48]. Overall, these neurodegenerative processes, characterized by the deposition of abnormal tau protein in the brain, are known as tau pathologies or tauopathies. They contribute to AD pathogenesis and are therefore also therapeutic targets in clinical studies for AD [4][40][4,45].3.4. Inflammation and Glial Responses
Under normal conditions, astrocytes and microglia, the brain´s main phagocytic immune cells, are essential to neuronal functioning and health. However, when the brain is injured, infected, or diseased, microglial cells are rapidly activated. They become highly movable, secreting inflammatory proteins, migrating to the affected area, and phagocytosing bacteria, aggregated proteins, cellular debris, and damaged neurons and synapses [44][45][52,53]. This intensive reaction of activated microglial cells is generally referred as microgliosis, which includes, in addition to their phagocytosing function, triggering inflammatory processes by releasing proinflammatory proteins. Among these proteins, small cytokine peptides and inflammasome-derived protein complexes are able to stimulate the production, deposition, and spreading of Aß in the brain [46][54]. Accordingly, cytokines, including some interleukins (ILs) as main triggers of inflammation, induce expression of the IFITM3 (interferon-induced transmembrane protein 3) protein in neurons and astrocytes. IFITM3 protein directly binds to γ-secretase and upregulates its activity, thereby increasing Aß production [47][55]. In contrast, pharmacological inhibition of the NLRP3 (nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3) inflammasome by the small molecule dapansutrile, led to reduced microglia activity and cortical amyloid plaque deposition, accompanied by improved cognitive abilities in AD mouse model [48][56]. The NLRP3 inflammasome is involved in processing cytokine precursors into active molecules. On the other hand, in an astrocyte-microglia cross-talk observed in AD human and mouse brain, interleukin-3 protein (IL-3) is released by astrocytes and activates the immune response of microglial cells, which then cluster around aggregates of Aß and tau and help to clear them [49][57]. This corresponds to the known function of microglia, which protects the brain by phagocytosing (engulfing and digesting) and thereby eliminating, e.g., aggregated proteins and unwanted or degenerating synapses [44][45][52,53]. In the activation of microglia, the gene triggering receptor expressed on myeloid cells 2 (TREM2) has been found to be causally involved [50][51][58,59]. This gene encodes a key receptor protein on the surface of microglia in the central nervous system, associated with signaling. TREM2 is thought to mediate repression of inflammatory cytokine production or secretion, and to increase microglial functions in recognizing and removing Aß by phagocytosis. Thus, decrease in CBF and ischemia have been shown to upregulate expression of TREM2 and other phagocytosis-related genes as well as components of astrocytic phagocytosis, suggesting improved ability to remove Aß by glial cells under these conditions [43][50][48,58]. Accordingly, loss of TREM2 gene function led to enhanced amyloid seeding and AD risk [50][58]. In contrast, acute TREM2 reduction in AD mouse brain increased microglial phagocytosis, accompanied by slowing Aß plaque deposition [51][59].4. Role of Aß in Triggering Vascular Constriction and CAA in AD
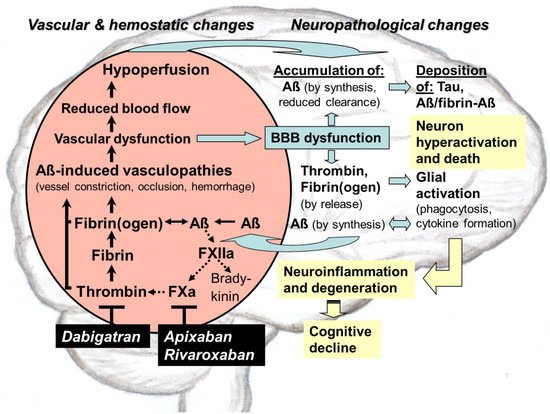
Cerebrovascular abnormalities, such as vascular lesions (e.g., hemorrhages, tissue injury), vessel occlusion (infarctions), cerebral small-vessel disease (CSVD), impaired vascular function and blood flow, are long-known and very early occurring, typical phenomena in AD. However, only in the last years, Aß-induced cerebrovascular constriction and damage to vessel walls, and their endothelial cells came into focus of therapeutic research [5][12][26][34][35][43][52][5,12,27,35,36,48,63]. The monolayer of endothelial cells, the endothelium, constitutes the inner lining of the vessel wall in contact with the blood and thus is part of the BBB. BBB is a special structure of brain vasculature that conveys selective and hemodynamically responsive movement of molecules between the blood and the brain [14][52][53][14,63,64]. Pathological deposition of Aß in the vessel wall, which proceeds along with the degeneration of smooth muscle cells, characterizes Aß-type CAA [5][26][33][34][35][36][5,27,34,35,36,37]. Together with early Aß-induced vascular constriction, CAA is a major cause for brain vasculopathies, leading eventually to vascular and BBB dysfunction in AD [26][43][27,48]. Moreover, CAA is the most prominent example of crosstalk between vascular and neuronal damage in AD [12][26][12,27] (Figure 1). Intriguingly, cerebrovascular amyloid was the starting material for the first isolation and characterization of Aß from AD brain [22][23].Please include review text section 4.5 in Biomedicines p12, as:
4.1. Pathophysiological Impact
The CAA-associated vasculopathies.....
4.1. AD Mouse Models
4.2. Occurrence of CAA
Cerebral amyloid angiopathies are commonly observed in the elderly brain and are classified into different types, according to the amyloid protein involved [26][56]. Among these classes, Aß-type cerebral amyloid angiopathy (CAA) is characterized by deposition of congophilic material consisting of Aß in meningeal and small to medium-sized cerebral blood vessels. With a prevalence of 82–98% [56], CAA is most commonly found in patients with sporadic, hereditary, or genetic AD. In the latter case, gene mutations are primarily associated with the Aß pathogenic syndrome [5][12][26][34][35]. CAA is particularly diagnosed in the neocortical and hippocampal brain areas, which correspond to the preferred locations of parenchymal Aß accumulation in AD. In CAA, mainly leptomeningeal and parenchymal small arteries, arterioles, capillaries, and, less frequently, veins are affected in their vascular activity and function through deposition of Aß aggregates in and around vessel walls [5][12][26][34][35][57]. Aß are first observed at the periphery of arterioles, the sites of initial Aß deposition and thus its seeding places [33][58][59]. Depending on CAA severity, Aß accumulation in the vessel wall displays a characteristic pattern with Aß initially deposited in the outer regions of the tunica media to the adventitia. Later, Aß accumulate in all layers of the small arteries and arterioles and can replace vessel wall often totally, except for the endothelial cells.4.3. Aß in CAA and Brain Parenchyma
Although Aß is the main component of parenchymal neuritic plaques, as well as of vascular deposits in CAA, the length of Aß involved appears to differ between these depositions [26][36]. Parenchymal Aß deposition in AD is composed mainly of Aß42, whereas in the vessel wall of CAA, the shorter subtype Aß40 is the predominant form. According to findings in AD mouse models, the deposition of parenchymal as well as of vascular Aß originates from a common neuronal source [26][36]. This indicates that neuron-derived Aß can migrate to and accumulate in the vasculature far from the site of generation. However, the underlying mechanism has not yet been elucidated in detail [36]. One hypothesis is that Aß42 is withheld in the parenchyma because of its lower solubility and, therefore, its tendency of forming insoluble plaques faster than the shorter Aß isoform. In contrast, the more soluble Aß40 does not aggregate so easily and diffuses more likely along perivascular drainage pathways across BBB into the blood system. In blood vessels of human AD brains, Aß40 accumulates and aggregates on vascular basement membranes, formed by endothelial cells and pericytes [26][36][58]. Thereby, Aß deposits are found first at the periphery of arterioles, alongside of putative ISF drainage routes [33][58][59].4.4. Brain Vasculopathies and Lesions by Aß-Driven CAA
In the brains of AD patients, CAA-associated vasculopathies lead to the development of hemorrhagic lesions (e.g., lobar intracerebral macrohemorrhage, cortical microhemorrhage, cortical superficial siderosis) and ischemic lesions (e.g., cortical microinfarcts, ischemic changes of the white matter) [26]. Moreover, encephalopathies are associated with CAA-related inflammation [26]. Empirically, CAA is accompanied by multiple or recurrent cerebral microhemorrhages, such as asymptomatic microbleeds and symptomatic, atypical bleeding [56]. The occurrence of new hemorrhages occurs preferentially at the sites of increasing amyloid deposition [26]. In addition, obliterating vascular changes leads to ischemic (micro-)infarcts or lacunes [56], which preferentially affect other cerebral vessels than those that rupture, causing cerebral hemorrhages. In cerebral blood vessels, close correlation has been found between the severity of Aß load and the impairment of vascular function. This dysfunction is expressed in reduced CBF, hypoperfusion (ischemia), and undersupply of the brain, particularly with oxygen (hypoxia) and glucose, a key energy substrate for the tissue [43][60][61][62][63] (Figure 1).4.5. Pathophysiological Impact of Aß on Vascular and BBB Functioning
The CAA-associated vasculopathies and the resulting vascular lesions entail pathophysiological consequences for the Aß-loaded, diseased brain that manifest themselves particularly in the disruption of vascular and BBB functioning [26][43][53][61][62][64] (Figure 1). This dysfunction of the cerebrovascular system is particularly detrimental because it obstructs cerebral blood flow (CBF) and thereby the perfusion of the brain. Full brain perfusion is indispensable to sustain normal tissue metabolism and neuronal functioning. Hypoperfusion restricts the supply of the brain tissue with a variety of vital constituents from the blood, including gas exchange of oxygen and CO2, ions and water, solutes (e.g., glucose and other carbohydrates, fatty acids, amino acids, hormones, vitamins, organic anions and cations, nucleotides), peptides and proteins, as well as cellular components [53][58][61][62][64][65].5. Interaction of Aß with the Plasma Contact System and its Driven Pathways of Coagulation and Inflammation in AD
5.1. Aß-Induced Activation of FXII in Contact System and Effects on Pathways Beyond
5.2. Pathological Dimension
The recently detected crosstalk between Aß and FXII, which stimulates production of thrombin, fibrin, fibrin-Aß clots, and bradykinin, as well as associated glial responses, is an important pathological feature, contributing to cerebrovascular and neuronal dysfunction in AD [5][13][5,13] (Figure 1). In brain parenchyma, accumulations of thrombin, fibrin(ogen), and Aß generate together a chronic inflammatory milieu [13]. Particularly, glial cells are activated and produce inflammatory cytokines, which elicit continued formation of Aß and spread of their aggregates in the brain. Cerebrovascular dysfunction is caused early by capillary constriction and occlusion, mediated by Aß, fibrin–Aß clots, platelet aggregation, and neutrophil trapping, as well as by developing CAA-related arterial damage, infarctions, and hemorrhage [5][13][43][5,13,48]. The resulting BBB breakdown and dysfunction leads to extravasation of thrombin and fibrin(ogen) from the blood into the brain parenchyma but also, vice versa, to reduced perivascular clearance of parenchymal Aß in the blood stream [53][64]. All effects together cause the collapse of CBF and brain perfusion, resulting in deficiency of oxygen and nutrients and a drop in the metabolism of the cerebral tissue [5][12][34][35][43][5,12,35,36,48]. As consequence, a self-amplifying spiral of increasing accumulation of Aß, thrombin, and fibrin(ogen) is elicited in the parenchyma, which is accompanied by enhanced formation and deposition of fibrin–Aß aggregates and tau pathologies. This further intensifies neuroinflammatory and degenerative processes as well as cerebrovascular damage in AD pathogenesis (Figure 1). Ultimately, loss of synapses and neurons cause memory and cognitive abilities to gradually disappear [5][12][34][35][43][5,12,35,36,48].6. Therapeutical Intervention Using Thrombin-Inhibiting Anticoagulants against Dysregulated Intrinsic Coagulation in AD
From the theoretical view, there are serious arguments for including the pathway of intrinsic coagulation as a therapeutic target in the strategy to treat AD. In the center of this approach is to address the harmful cerebrovascular changes in AD. These changes lead to the collapse of the vascular and BBB function and of the blood supply for the nervous system parenchyma, causing neuroinflammatory and neurodegenerative responses. An important trigger of this disastrous vascular scenario is Aß-driven, out-of-control production of thrombin in the blood. This thrombin overproduction leads to increasing formation of fibrin and degradation-resistant fibrin–Aß clots, as well as to promoted platelet aggregation and inflammation (Figure 1). Therapeutic intervention into this vicious circle focuses on directly blocking activity or production of thrombin by treatment with anticoagulants. As result, a variety of adverse processes in AD might be decelerated, stopped, or prevented (Figure 1). These include (i) the deposition of fibrin–Aß clots and oligomeric Aß in cerebral vessels, leading to vascular and BBB dysfunction; (ii) reductions in CBF, brain perfusion, and nutrient supply; (iii) the accumulation of extravasated, inflammatory thrombin and fibrin(ogen) in the brain parenchyma; (iv) the accumulation and aggregate spreading of parenchymal Aß by hypoxia- and glia-induced Aß synthesis, as well as by impaired perivascular Aß clearance due to BBB dysfunction; (v) glial activation and associated neuroinflammatory responses; (vi) Aß-induced tau pathologies; (vii) neuronal damage with loss of synapses and neurons, leading to cognitive impairment. Consequently, a thrombin-inhibiting treatment might offer a therapeutic chance to counteract Aß-induced vasculopathies and dysfunctions in AD, and to prevent hypoperfusion, hypoxia, and nutrient deficits, as well as neuroinflammatory milieus in the brain, elicited by accumulating Aß, thrombin, and fibrin(ogen). As a possible therapeutic success, neuroinflammatory and neurodegenerative alterations, leading to memory and cognitive decline, could be slowed down, stopped, or prevented. However, the therapy of patients should be initiated as early as cognitive deficits by AD are suspected and clinically confirmed. As a precaution, patients with diagnosable CAA should be excluded from the therapy to avoid bleeding risk [9][10][11][13][70][9,10,11,13,102]. The first preclinical study for a detailed proof-of-concept has been carried out by Cortes-Canteli and co-workers [71][93]. In order to prevent harmful effects of thrombin in AD mouse brains, the authors employed long-term anticoagulation with dabigatran, which directly blocks thrombin activity [71][93]. The results revealed that dabigatran is able to inhibit the formation of occlusive fibrin thrombi in cerebral vessels, decreases in CBF, brain hypoperfusion, and memory decline [71][93]. Concomitantly, dabigatran treatment preserved BBB function, proved by pericyte integrity and the absence of AD-related astrogliosis [71][93]. Likewise, dabigatran treatment significantly decreased amyloid plaque deposition as well as halved the accumulation of surrounding Aß oligomers [71][93]. Concomitantly, the neuroinflammatory milieu in the brain tissue was reduced, demonstrated by lowered amounts of phagocytic microglia and infiltrated peripheral T cells [71][93]. In addition, no hemorrhages or incidents of intracerebral bleeding were observed [71][93]. These findings highly confirm conclusions, drawn from basic research, on possible benefits of a thrombin-inhibiting therapy in AD [9][10][11][13][70][71][72][9,10,11,13,93,102,118] (Figure 1).Please add modified sentences in text Biomedicines p20, from 6.3.1. and 6.3.2.
The first clinical studies on anticoagulant medication against dementia in small groups of senile-presenile patients extend far back into the 1960s and resulted in decelerated cognitive decline and reduced morbidity and mortality [73][74][75][76][119-122]. In addition, over the last 20 years, multiple observer studies and systematic reviews on patients with anticoagulant use due to atrial fibrillation (AF) suggest that oral anticoagulants, particularly DOACs, safeguard against incidence of dementia like AD, as shown in persons without a dementia history before treatment [77][78][79][80][81][82][83][84][123-130].
7. Clinical Perspective for Anticoagulant Use against AD
7.1. Evaluation of Therapeutic Suitability of Available Anticoagulants
The parenteral administration of a drug by injection or infusion directly into the blood stream are less suitable for a permanent therapy from their handling alone. This would be the case when treating parenteral, indirect thrombin-inhibiting heparins (e.g., enoxaparin), heparinoid danaparoid sodium, and fondaparinux or direct thrombin-inhibiting hirudin, bivalirudin, and the synthetic L-arginine derivative argatroban. Heparins, hirudin, and derivatives are usually administered for short-term prophylaxis of thromboembolic events and for therapy of acute venous thrombosis [16]. Undesirable side effects of treatment with heparins are given by abnormally low levels of platelets in the blood (thrombocytopenia) and increased risk of bleeding. In addition, anticoagulation by heparins is unpredictably affected by unspecific plasma protein binding. Furthermore, heparins do not inhibit fibrin-bound thrombin and related thrombus formation [16][85][16,133]. In contrast to heparins, hirudin inactivates thrombin bound to clots and does not directly interact with platelets. However, in therapeutic application, bleeding complications have been frequently observed [86][134].
With respect of administration and handling, oral treatment with anticoagulants for therapy would certainly be preferable to a parenteral application, if the efficacy and safety profile of the oral drug is also favorable and the patient can take the medication on a consistent basis. For oral administration, anticoagulants are available on the market from VKA-type, such as warfarin, phenprocoumon, as well as from DOAC-type with the direct thrombin inhibitor dabigatran and the direct FXa inhibitors apixaban, betrixaban, edoxaban, rivaroxaban [16]. Dabigatran, rivaroxaban, and apixaban were approved for antithrombotic use already at the beginning of the 2010s years [16]. Approval of edoxaban and, subsequently, betrixaban was in the second half of the decade [16]. The development of the new class of antithrombotic DOACs that directly target to specific factors in intrinsic coagulation was particularly desired due to serious disadvantages of the conventional VKAs [16][19][87][16,20,136].
Compared to VKAs, DOACs provide constant therapeutic efficacy and a more favorable safety profile, as well as avoidance of adverse effects from vitamin K deficiency due to their different mechanism of action. In detail, the advantages of DOACs include (i) rapid onset of action, (ii) short half-life, (iii) less drug–drug interactions and no dietary interactions, and (iv) safe antidote strategies in situations of bleeding risk. Therefore, the lower intra- and interindividual variability in the DOAC-effect allows fixed dosing and a predictable anticoagulative response without the need for continuous monitoring of the drug level in patients [9][11][16][19][87][88][89][9,11,16,20,136,138,139]. Since DOACs, especially dabigatran, are eliminated to a large extent via the kidney, the renal function in patients should be routinely monitored, particularly in elderly persons due to increasing renal impairment and associated co-morbidities [19][87][20,136]. In patients with renal impairment, dependent on severity, DOAC use requires dose adjustments or is contraindicated [19][87][20,136]. In addition, DOAC-type anticoagulants also hold the risk of bleeding, particularly of serious intracranial hemorrhage [16][19][87][16,20,136].
In a systematic review and meta-analysis of phase III trials for stroke and systemic embolism prevention in patients with AF (2009–2013), DOACs (apixaban, dabigatran, edoxaban, rivaroxaban) showed a more favorable risk–benefit profile compared with warfarin [90][140]. In addition, reductions in all-cause mortality and systemic embolic events, these agents reduced the risk of hemorrhagic stroke by 51% and the risk of intracranial hemorrhage by 52% [90][140]. This favorable efficacy and safety profile was consistent with many subgroups and ethnicities including the Asian population [87][90][136,140]. Conversely, dose-dependently, the risk of gastrointestinal bleeding was 25% higher with DOACs than with warfarin [90][140]. Similar results were obtained in a large retrospective observational study of ca. 400,000 AF patients, based on US claims data (2013–2015) [89][139]. Treatment with DOACs (dabigatran, apixaban, rivaroxaban) was associated with lower rates of stroke and systemic embolism, compared with warfarin. In addition, apixaban and dabigatran showed lower rates of major bleeding, including gastrointestinal bleeding, intracranial hemorrhage, and major bleeding at other key sites, whereas rivaroxaban had a higher rate of major bleeding, compared with warfarin [89][139]. This is in accordance with results from a new-user retrospective cohort study of patients with AF and dementia (2011–2017), comparing DOAC treatment versus warfarin [91][141]. DOAC-treated patients, who were older and had more comorbidities than the warfarin-treated individuals, showed similar prevention of thromboembolic events, compared to warfarin, but a reduced risk of intracranial bleeding [91][141]. However, the risk of gastrointestinal bleeding was increased in the DOAC treatment [91][141].
7.2. DOAC-Type Anticoagulants for In-Depth Clinical Investigation
Altogether, the wide range of clinical observer studies, conducted in the last 15 years particularly in elderly individuals with AF, demonstrate that DOAC-type anticoagulants exhibit a predictable therapeutic effect in preventing stroke and systemic embolic events. In addition, DOACs show a safety profile that more than halves the risk of dangerous intracranial hemorrhage in elderly people compared to VKAs [87][90][92][136,140,143]. These properties, along with their pharmacological advantages, give DOACs a clear preference over VKAs, particularly when anticoagulants are administered to elderly people, who are more vulnerable individuals. In addition, gastrointestinal bleedings, which are more likely to be promoted by DOACs compared to VKAs [87][90][91][136,140,141], can be treated better and stopped immediately after occurrence by effective antidote strategies. These strategies have been successfully developed for various DOACs in the recent years [16]. In the case of dabigatran, the specific antidote idarucizumab (Praxbind®) was introduced in 2016 [93][147]. This antibody binds to dabigatran with high affinity and leads within minutes to a rapid cancellation of the anticoagulative effect, e.g., in emergency operations or in situations of uncontrollable bleeding [16][93][16,147]. In the case of the FXa inhibitors apixaban and rivaroxaban, andexanet alfa (AndexXa®) was recently approved as a fast-acting antidote [16][94][16,148]. Andexanet alfa is a recombinantly modified, human FXa molecule, which itself has no effect on blood clotting. It acts as a kind of decoy protein that binds the FXa-inhibitors and thus restores blood clotting [16][94][16,148]. Generally, availability of an efficient and fast acting reversal agent should be a prerequisite for a long-term anticoagulative treatment. This is especially the case in elderly and comorbid AD patients, showing inherent bleeding risk due to fragile blood vessels [57][68].