Prostate cancer has become the second most common cancer and the fifth most deadly cancer in the world
[1]. In the United States, prostate cancer is the second leading cause of cancer-related death in men, projected to have 268,490 new cases and 34,500 deaths in 2022
[2]. Although localized prostate cancer may be successfully treated with radical prostatectomy or radiotherapy
[3], standard androgen deprivation therapy (ADT) followed by chemotherapy (Taxanes), androgen synthesis inhibitor (abiraterone), or androgen receptor (AR) antagonists (enzalutamide, apalutamide, and darolutamide) remains the primary treatment option for patients with advanced prostate cancer
[4]. Unfortunately, despite initial response to the current therapy, all patients eventually will develop from castration-resistant prostate cancer (CRPC) to drug-resistant CRPC through androgen-dependent or androgen-independent mechanisms. Androgen-dependent mechanisms include a rise in AR splice variants
[5][6], AR overexpression
[7], intratumoral or alternative androgen biosynthesis
[8], and AR mutations
[9], while androgen-independent mechanisms are mediated through the activation of DNA repair pathways
[10], PI3K/AKT/mTOR pathways
[11][12], BRAF-MAPK
[13], Wnt signaling pathways
[14][15], glucocorticoid receptor pathways
[16], and neuroendocrine differentiation
[17]. Multiple clinical trials are exploring therapeutic strategies to overcome these resistance mechanisms, including those utilizing immunotherapies that work in conjunction with the patient’s immune system.
Harnessing the patient’s immune system to fight tumors has reinvigorated the field of cancer therapy over the past few decades. Tumor immunotherapy has ushered in a new era of cancer treatment, bringing hope to patients with advanced stages of cancers, including prostate cancer
[18]. In fact, immune checkpoint inhibitors targeting programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) and CTLA-4 (both of which suppress T-cell proliferation) have shown unprecedented, sustained responses in certain tumor types. These cancers (e.g., melanoma
[19], kidney cancer
[20], and non-small-cell lung cancer
[21]) are often referred to as having “hot tumors” because of the considerable reliance they have on immune checkpoints for propagation. In contrast, “cold tumors”, such as prostate cancer, have low tumor immunogenicity
[22] and an active immunosuppressive tumor microenvironment (TME)
[23], resulting in a very limited response to immunotherapy. The majority of prostate tumors are identified as immune-ignorant, typically characterized by decreased levels of antigen-expressing molecules
[24], the low expression of genes involved in antigen processing and presentation, and deficient cytotoxic T-cell recruitment and activation
[25][26]. These factors contribute to prostate tumors being “cold”, since they are less responsive to immune factors as other cancers. This leads to difficulties when testing immunotherapies, such as immune checkpoint inhibitors in prostate cancer. A better understanding of the molecular mechanisms that lead to immunosuppression in prostate cancer and the development of better treatment options will help address the current clinical dilemma in prostate cancer immunotherapy.
2. The Development of Prostate Cancer Immunotherapy
2.1. Sipuleucel-T
Sipuleucel-T is an active cellular immunotherapy that stimulates T-cell immune responses and is currently the only anticancer vaccine approved for the treatment of asymptomatic or mildly symptomatic metastatic CRPC (mCRPC) patients. It consists of autologous peripheral blood mononuclear cells (PBMCs) that are activated in vitro with a recombinant fusion protein (PA2024) containing the prostate antigen, prostatic acid phosphatase, and granulocyte-macrophage colony stimulating factor (GM-CSF) to expand antigen-loaded antigen-presenting cells (APCs) and then reinfuse back to patients after 36–44 h of in vitro culturing to sensitize/activate the patient’s own T cells
[27]. Compared with a placebo, patients in the Sipuleucel-T arm had a median survival of 4.1–4.5 months longer
[27][28] and a 22–33% lower risk of death
[29]. In addition, preliminary safety and efficacy data for the combination of Sipuleucel-T with AR-targeting agents
[30], PD-L1 inhibitor atezolizumab
[31], or Radium-223
[32] in mCRPC patients are still in development. Further studies in larger trials are necessary to verify the positive effects of these combination therapies. Sipuleucel-T is a landmark of prostate cancer immunotherapy. However, the modest efficacy and economically low cost-effectiveness restrict its application. Several other prostate cancer vaccines, including PROSTVAC/PCa and Anti-RhoC, have been tested in clinical trials (
Table 1).
Table 1. Completed vaccine and immune checkpoint inhibitor clinical trials for CRPC.
| Clinical Trial Number and Trial Phase |
Description |
Results (OS in Months; PSA in ng/mL) |
Ref. |
| Sipuleucel-T (NCT00065442) |
III |
Active cellular (peripheral-blood mononuclear and antigen-presenting cells) |
OS, 25.8 with Spiuleucel-T 21.7 with placebo; PSA, 51.7 with Sipuleucel-T 47.2 with placebo |
[27] |
| PROSTVAC (EudraCT 2010-021196-85) |
III |
Recombinant vaccinia and fowlpox viruses containing transgenes for human PSA and 3 T-cell costimulatory molecules |
OS, 23.1 with viral vectors 22.8 with placebo; PSA, 71.4 with viral vectors 82.6 with placebo |
[33] |
| VANCE (NCT02390063) |
I |
Replication-deficient viruses targeting oncofetal self-antigen 5T4 (early-stage PCa) |
PSA, >100% increase in PSA levels post-vaccination for 3 participants, with others showing <50% increase |
[34] |
| KRM-20 (UMIN000011028) |
II |
KRM-20 is a 20 peptide mix that induces cytotoxic T-lymphocytes against 12 tumor-associated antigens |
No significant difference in PSA response, but both human leukocyte antigen (HLA)-IgG and CTL responses increased in KRM-20 arm |
[35] |
| Blood-derived dendritic cells (DCs) (NCT02692976) |
IIa |
Monotherapies or combinations of myeloid DCs and/or plasmacytoid DCs used to induce cytotoxic T cells (intranodal injection) |
Radiographic progression-free survival (rPFS) found to be 18.8 months in those with functional antigen-specific T cells (n = 5), and 5.1 months in those without (n = 16) |
[36] |
| Anti-RhoC (NCT03199872) |
I/II |
The RhoC protein has been correlated with advanced cancer cells and metastasis, so this trial tests a vaccine to inhibit its function |
86% of patients had a significant T-cell response during vaccinations, and 90% during the follow-up (functional T effector memory cells were seen, but not Tregs) |
[37] |
| PCD4989g Atezolizumab (NCT01375842) |
I |
Small-molecule atezolizumab (PD-L1 inhibitor) with previous treatment using Sipuleucel-T or enzalutamide |
PSA, 8.6% response, OS, 14.7 months, overall limited efficacy, so combination approach may be needed |
[38] |
| IMbassador250 Atezolizumab (NCT03016312) |
III |
Small-molecule atezolizumab (PD-L1 inhibitor) with previous treatment using abiraterone; concurrent with enzalutamide for both arms |
Stopped early because patients were at risk of immune-mediated adverse events; OS, 15.2 months for atezolizumab + enzalutamide vs. 16.6 months for enzalutamide only |
[39] |
| KEYNOTE-199 Pembrolizumab (NCT02787005) |
II |
Monoclonal antibody pembrolizumab (PD-1 inhibitor) with previous treatment using docetaxel or enzalutamide |
PSA, <10% response, ORR, <5%, rPFS, 2.1, 2.1, and 3.7 months for 3 cohorts (Cohort 1: PD-L1-positive; Cohort 2: PD-L1-negative; Cohort 3: bone-predominant disease, regardless of PD-L1 expression) |
[40] |
| STARVE-PC Ipilimumab/Nivolumab (NCT02601014) |
II |
Ipilimumab (anti-CTLA4 monoclonal antibody), nivolumab (PD1 inhibitor), some concurrent treatment with nivolumab (all with enzalutamide) |
Lower alkaline phosphatase levels in a subset of patients treated with immune blockade; did not meet primary endpoint |
[41] |
| CheckMate650 Ipilimumab (NCT02985957) |
II |
Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor); one subset treated with cabazitaxe. Concurrent with nivolumab and higher dose ipilimumab |
OS, 15.2 months in post-chemo cohort and 19 months in pre-chemo; ORR, 10% in post-chemo cohort and 25% in pre-chemo |
[42] |
| MDX-010 Ipilimumab (NCT00323882) |
I/II |
Ipilimumab (CTLA4 inhibitor), dose-escalation treatments of ipilimumab combined with radiotherapy |
PSA, 8 patients had PSA decline >50% and 1 had a complete response; high dose of 10 mg/kg ipilimumab showed a manageable safety profile |
[43] |
2.2. Adoptive Immune Cell Immunotherapy
Adoptive cell therapy, which utilizes autologous activated immune effector cells to eradicate tumor cells, has achieved promising results in a variety of hematological malignancies
[44]. However, as they lack specific tumor antigens and due to the existence of an immunosuppressive TME in solid tumors and the expression of inhibitory molecules (such as PD-1) by tumor cells, the activity and proliferation of T cells are always suppressed. Additionally, the abilities of T cells to recognize and kill tumor cells are impaired, resulting in the limited application of chimeric antigen receptor-T (CAR-T) in solid tumors
[45]. In prostate cancer, autologous T lymphocytes collected by peripheral blood leukapheresis can be genetically engineered to express CARs that recognize specific prostate tumor antigens and obtain specific immune effector cells. CAR-T cells identify and infiltrate tumor cells by targeting surface antigens without the presence of APCs. There have been five generations of CARs, and their molecular structures and physical properties significantly alter CAR-T cells’ effects in therapeutics
[46]. Currently, PSMA-CAR-T cells (NCT04429451 and NCT04249947), PSCA-CAR-T cells (NCT03873805), and CAR-T-PSMA-TGFβRDN69 cells (NCT03089203 and NCT04227275) are highly effective in mCRPC tested in phase I or phase II clinical trials
[37][47][48][49][50]. Natural killer (NK) cells can also be engineered to express the CAR, and recruitment is currently ongoing for an early phase I clinical trial evaluating anti-PSMA CAR-NK cells (NCT03692663)
[51].
2.3. Immune Checkpoint Inhibitors
The advent of immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway has reinvigorated the treatment of various advanced cancers
[52]. However, the stronger immunosuppressive microenvironment renders prostate cancer to be less susceptible to immune checkpoint blockades. Studies have illustrated that PD-1/PD-L1 inhibitors provide suboptimal clinical benefit in an unselected population of patients with mCRPC
[38][53]. Nonetheless, long-term survival benefits and sustained complete responses have been reported in some mCRPC patients receiving a CTLA-4 inhibitor (Ipilimumab)
[54], suggesting that selected prostate cancer patients may benefit from a checkpoint blockade. Accordingly, a more widely accepted explanation for the suboptimal clinical activity is that the majority of mCRPC patients have an immunosuppressive TME with the low infiltration of CD8+ T cells and an increased influx of immunosuppressive cells. Therefore, clinical trials designed for combining anti-PD-1/PD-L1 agents with other anticancer treatments, including inhibitors of different immune checkpoint pathways or other systemic anticancer treatments, mostly failed in prostate cancer at the current stage (
Table 1).
In a phase Ia study using atezolizumab in patients with mCRPC which progressed after Sipuleucel-T or enzalutamide treatment, 8.6% of patients had a 50% PSA response, and only one patient had an objective partial response, with a median overall survival (OS) of 14.7 months
[38]. In the phase II KEYNOTE-199 trial, pembrolizumab had less than 10% of the PSA response rate, less than 5% of an overall response rate (ORR), and 2.1, 2.1, and 3.7 months of radiographic PFS (rPFS) for the three cohorts, respectively (Cohort 1: PD-L1-positive; Cohort 2: PD-L1-negative; Cohort 3: bone-predominant disease, regardless of PD-L1 expression)
[40]. In addition, a phase III trial (IMbassador250) showed that the addition of the PD-L1 inhibitor atezolizumab did not increase the efficacy of enzalutamide in mCRPC patients whose disease had progressed on abiraterone (
n = 759)
[39]. The OS endpoints were not reached, and the trial was terminated. From the trial, longer PFS was associated with the addition of atezolizumab to enzalutamide in men selected for PD-L1 immune checkpoint expression and high levels of CD8+ T cells. Emerging data suggested that AR is a negative regulator of CD8+ T cells in responding to anti-PD1/PD-L1 treatment
[55]. AR status in T cells may act as an important efficacy predictor to the immune checkpoint inhibitors, and complete androgen axis blockage is critical to achieve the best antitumor results by immune checkpoint inhibitors in CRPC patients. Thus, the potential synergy between atezolizumab and enzalutamide might be useful in certain selected patients and should be verified by future clinical trials
[56].
Ipilimumab, a popular inhibitor of the immune checkpoint molecule CTLA-4, was used to treat in prostate cancer patients after it was approved by the FDA to treat melanoma. Ipilimumab, when taken as a monotherapy or combination treatment in phase I/II clinical trials of mCRPC, did not exacerbate immune-related adverse effects
[57]. The combination of ipilimumab and radiotherapy improved OS (2- to 3-fold higher) in patients with mCRPC after docetaxel, while combination therapy with the PD-1 inhibitor nivolumab showed only modest activity in AR-V7-positive mCRPC patients
[41]. The ongoing phase II trial of nivolumab in combination with ipilimumab (CheckMate 650) divides patients with mCRPC before and after chemotherapy into two cohorts. A preliminary analysis on 90 treated patients showed objective response rates of 25% and 10%, respectively, with a median OS of 19 and 15.2 months before and after chemotherapy; two patients in each cohort had complete responses
[42].
Despite the unfavorable results in prostate cancer by current immune checkpoint inhibitors, PD-1 inhibitors including nivolumab and pembrolizumab, anti-PD-L1 inhibitors including atezolizumab and avelumab, and anti-CTLA4 inhibitor ipilimumab
[58] continue to be rigorously tested in multiple clinical trials in CRPC patients (
Table 2). The ongoing clinical trials for immune checkpoint inhibitors in prostate cancer will further examine the applicability of these inhibitors to clarify whether they may improve treatment outcomes for patients with CRPC either alone or in combination with other treatment strategies.
Table 2. Ongoing clinical trials utilizing immune checkpoint inhibitors in CRPC patients.
| Trial Name and Trial Phase |
Treatment(s) |
Purpose and Expected Completion Date |
| CHOMP (NCT04104893) |
II |
Pembrolizumab (PD-1 inhibitor) |
To evaluate the activity and efficacy of pembrolizumab in mismatch repair deficiency (dMMR) and CDK12 biallelic inactivation mCPRC patients |
3/2023 |
| PERSEUS1 (NCT03506997) |
II |
Pembrolizumab (PD-1 inhibitor) |
To evaluate the efficacy of pembrolizumab. To determine PD-1 and PD-L1, Treg infiltration, CD3, CD8, and lymphocyte infiltration |
9/2025 |
| NCT03406858 |
II |
Pembrolizumab (PD-1 inhibitor), HER2Bi-armed activated T cells |
To test if the combination of the HER2Bi-armed T cells and pembrolizumab is better at treating mCRPC patients |
12/2021 (Active) |
| INSPIRE (NCT04717154) |
II |
Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor) |
To evaluate the effects of 4 cycles of combination treatments (ipilimumab and nivolumab), followed by monotherapy nivolumab in participants with mCPPC |
6/2025 |
| IMPACT (NCT03570619) |
II |
Ipilimumab (CTLA4 inhibitor), nivolumab (PD1 inhibitor) |
To evaluate the efficacy of combo treatment in patients with mCRPC and CDK12 mutations |
5/2023 |
| NCT03456804 |
II |
ESK981 (Pan-VEGFR/TIE2 tyrosine kinase inhibitor and PIKfyve lipid kinase inhibitor) |
To study the side effects and how well ESK981 works in treating patients with mCRPC |
10/2022 |
| NCT03792841 |
I |
Acapatamab (bispecific T-cell engager), pembrolizumab (PD-1 inhibitor) |
To determine the max tolerated dose of Acapatamab (a half-life extended (HLE) bispecific T-cell engager (BiTE®) construct) alone and in combination with pembrolizumab |
6/2025 |
| NCT05293496 |
I |
MGC018 (CD276 inhibitor), lorigerlimab (dual PD-1 × CTLA-4 inhibitors) |
To determine the safety and efficacy of MGC018 + lorigerlimab combo treatment |
3/2025 |
| NCT05177770 |
II |
SRF617 (CD39 inhibitor), etrumadenant (dual A2aR/A2bR antagonist), zimberelimab (PD-1 inhibitor) |
To evaluate the safety and efficacy of SRF617 in combination with etrumadenant and zimberelimab |
11/2023 |
| IceCAP (NCT03673787) |
I/II |
Ipatasertib (AKT inhibitor), atezolizumab (PD-L1 inhibitor) |
Proof of concept for the combination of ipatasertib and atezolizumab acting on PI3K hyperactivated tumors |
11/2023 |
| NCT03061539 |
II |
Nivolumab (PD1 inhibitor), ipilimumab (CTLA4 inhibitor) |
To evaluate the efficacy of PD-1 inhibitor in combination with CTLA4 inhibitor |
7/2025 |
| NCT02933255 |
I/II |
PROSTVAC-V/F (vaccine), nivolumab (PD1 inhibitor) |
To evaluate the combination therapy of PROSTVAC and nivolumab for safety and effectiveness |
8/2022 |
| Rad2Nivo (NCT04109729) |
Ib/II |
Nivolumab (PD1 inhibitor), radium-223 (radioactive isotope) |
To assess the safety of this combination treatment, then expand into a phase II cohort |
4/2025 |
| NCT04159896 |
II |
ESK981 (multi-tyrosine kinase inhibitors), nivolumab (PD1 inhibitor) |
To evaluate the safety and efficacy of these drugs in combination (ESK981 = pan-VEGFR/TIE2 tyrosine kinase inhibitor) |
3/2022 (Active) |
| NCT03651271 |
II |
Nivolumab (PD1 inhibitor), ipilimumab (CTLA4 inhibitor) |
To evaluate treatment outcomes for patients with low vs. high levels of CD8 cells in tumor biopsy in monotherapies of nivolumab or combo |
5/2023 |
| CheckMate 7DXNCT04100018 |
III |
Nivolumab (PD1 inhibitor), prednisone, docetaxel |
To assess the safety and efficacy of nivolumab + docetaxel in comparison to placebo + docetaxel |
8/2027 |
| NCT05169684 |
II |
BMS986218 (CTLA4 inhibitor), docetaxel, nivolumab (PD1 inhibitor) |
To assess the safety and efficacy of BMS986218 in different combos with nivolumab and docetaxel |
2/2026 |
PORTER
(NCT03835533) |
I |
NKTR-214 (CD122-preferential IL2 pathway agonist), nivolumab (PD1 inhibitor), SBRT (radiation), CDX-301 (FLT3 ligand, a dendritic cell mobilizer), INO-5151 (combination of DNA plasmids encoding IL-12 and PSA/PSMA) |
To evaluate the safety and efficacy of immunotherapy combinations. To explore immune biomarker response in prostate cancer after treatment with different combinations |
3/2023 |
| STELLAR-001(NCT03845166) |
I |
XL092 (tyrosine kinase inhibitor that targets VEGF receptors, c-Met), atezolizumab (PD-L1 inhibitor), avelumab (PD-L1 inhibitor) |
To evaluate the safety, tolerability, pharmacokinetics (PK), preliminary antitumor activity by XL092 as a monotherapy or in combination with other PD-L1 inhibitors |
11/2024 |
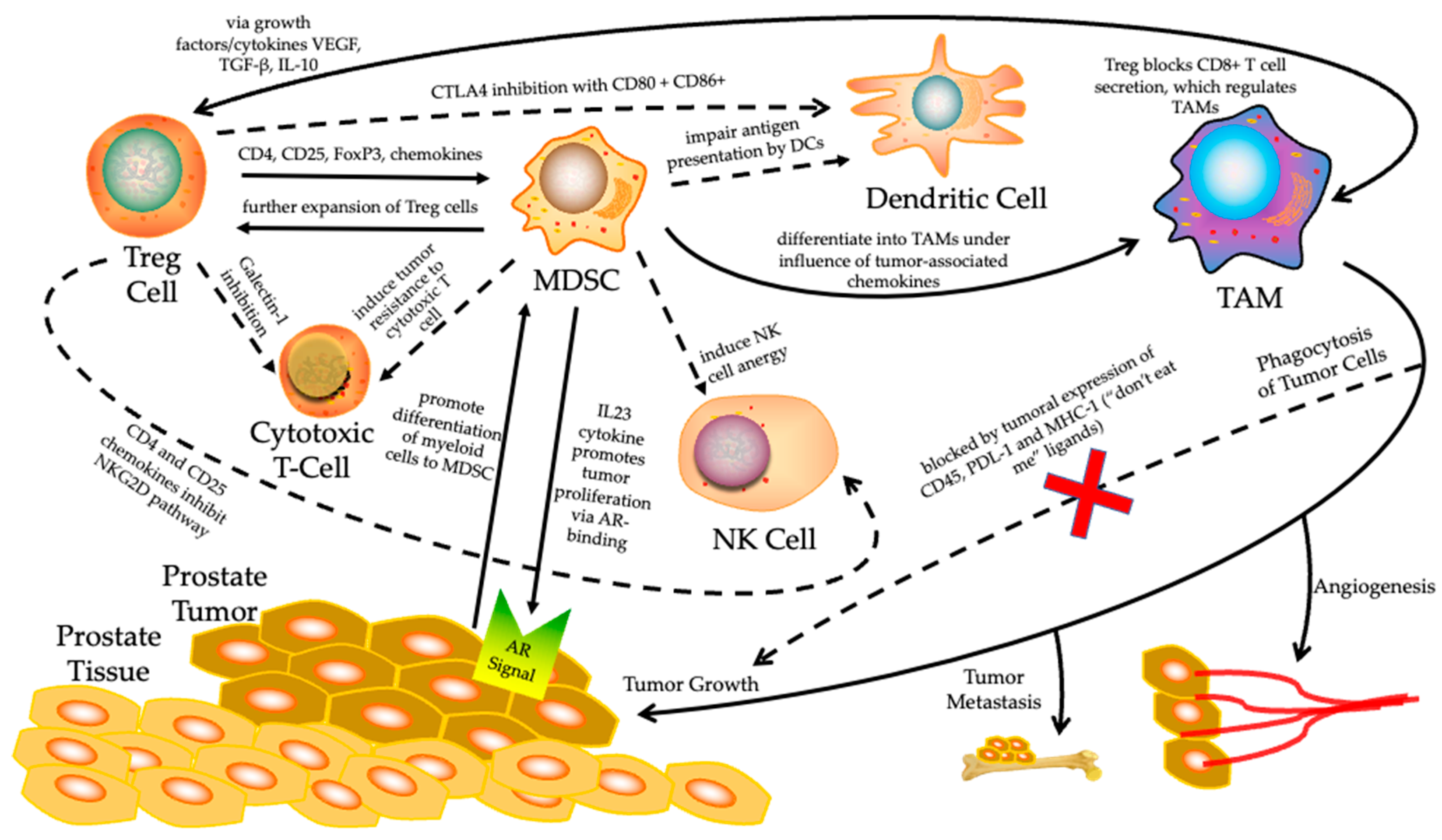
3. Role of TME in Therapy-Resistant Prostate Cancer Immune Evasion
Prostate cancer is considered a heterogeneous disease with a highly complex TME. Encompassed by a non-inflammatory TME and the low expression of neoantigens which would signal unchecked cell growth, prostate tumors have difficulty being differentiated by the immune effector system. Meanwhile, prostate tumors tend to evade antitumor immune cells through the secretion of immunosuppressive factors such as interleukin 1 beta (IL-1β), IL-10, and TGF-β in the TME and thereby induce the differentiation of myeloid cells into myeloid-derived suppressor cells (MDSCs). Concurrently, the increased infiltration of regulatory T cells (Tregs) and tumor-associated macrophages, in combination with decreased circulating NK cells in prostate cancer tissues, are associated with worse prognosis and resistance to immunotherapy
[59] (
Figure 1).
Figure 1. The tumor immune microenvironment (TIME) surrounding prostate cancer. It is known to be highly immunosuppressive, utilizing many cellular pathways which inhibit normal immune function and promote a tumor’s unchecked proliferation. Solid arrows represent upregulation of that cell’s expression or a specific function at the end of the arrow. Dashed arrows indicate inhibition of the cell or function at the end of the arrow. Specific mechanisms by which upregulation or inhibition work are summarized on their respective arrows. Treg, regulatory T; MDSC, myeloid-derived suppressor cell; NK, natural killer; TAM, tumor-associated macrophage; DC, dendritic cell; AR, androgen receptor.
Regulation of Tumor Immune Microenvironment (TIME)
In order to promote tumor growth, the TIME consists of tumors and their surrounding blood vessels, extracellular matrixes (ECMs), fibroblasts, immune cells, bone marrow-derived inflammatory cells, various signaling molecules, and other components, which play crucial roles in the antitumor immune response
[60]. T-cell composition, vascular networks, and cytokine diversity in the TIME are determinants of T-cell-mediated antitumor responses. There is complex interplay between the stromal cell background of fibroblast infiltration, the metabolic state promoted by the disturbed vasculature, and the subsequent hypoxia leading to an immunosuppressive TIME
[61]. Cytokines such as VEGF, TGF-β, and IL-10 are responsible for the recruitment of Tregs in the TIME and inhibit the proliferation, activation, and infiltration of cytotoxic lymphocytes. Therefore, the TIME plays a key role in prostate cancer progression and immune evasion. Previous studies suggest that the presence of tumor-infiltrating lymphocytes (TILs) is generally associated with better prognosis
[62][63]. However, prostate cancer TIL populations are mainly composed of CD4+FOXP3+CD25+ Treg cells and M2-type tumor-associated macrophage (TAM) cells, which contribute to the production of inhibitory cytokines and the maintenance of self-tolerance to suppress the immune response
[64]. They are associated with a higher risk of metastatic disease at diagnosis and worse distant metastasis-free survival
[65]. In addition, chemokines (such as CCL22) and chemokine receptors (such as CXCR4 and CXCR5) expressed by Tregs and MDSCs also contribute to immunosuppressive TIME formation in prostate cancer
[66].
 +1 credit
+1 credit