Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yuan, J.; Zhang, Q.; Chen, S.; Yan, M.; Yue, L. LC3-Associated Phagocytosis in Bacterial Infection. Encyclopedia. Available online: https://encyclopedia.pub/entry/26006 (accessed on 08 June 2026).
Yuan J, Zhang Q, Chen S, Yan M, Yue L. LC3-Associated Phagocytosis in Bacterial Infection. Encyclopedia. Available at: https://encyclopedia.pub/entry/26006. Accessed June 08, 2026.
Yuan, Jin, Qiuyu Zhang, Shihua Chen, Min Yan, Lei Yue. "LC3-Associated Phagocytosis in Bacterial Infection" Encyclopedia, https://encyclopedia.pub/entry/26006 (accessed June 08, 2026).
Yuan, J., Zhang, Q., Chen, S., Yan, M., & Yue, L. (2022, August 10). LC3-Associated Phagocytosis in Bacterial Infection. In Encyclopedia. https://encyclopedia.pub/entry/26006
Yuan, Jin, et al. "LC3-Associated Phagocytosis in Bacterial Infection." Encyclopedia. Web. 10 August, 2022.
Copy Citation
LC3-associated phagocytosis (LAP) is a noncanonical autophagy process reported in recent years and is one of the effective mechanisms of host defense against bacterial infection. During LAP, bacteria are recognized by pattern recognition receptors (PRRs), enter the body, and then recruit LC3 onto a single-membrane phagosome to form a LAPosome. LC3 conjugation can promote the fusion of the LAPosomes with lysosomes, resulting in their maturation into phagolysosomes, which can effectively kill the identified pathogens.
LC3-associated phagocytosis
bacterial infection
phagocyte
1. Introduction
The innate immune system is the first line of defense against the invasion of pathogenic microorganisms in the host. When they invade, the body can engulf, hydrolyze, and clear them, and manufacture the corresponding epitopes for activating the body’s immune response to the infection [1]. LC3-associated phagocytosis (LAP) plays a very important role in clearing pathogenic microbial infections, and its mechanism is different from phagocytosis and canonical autophagy (hereafter autophagy) (Figure 1).
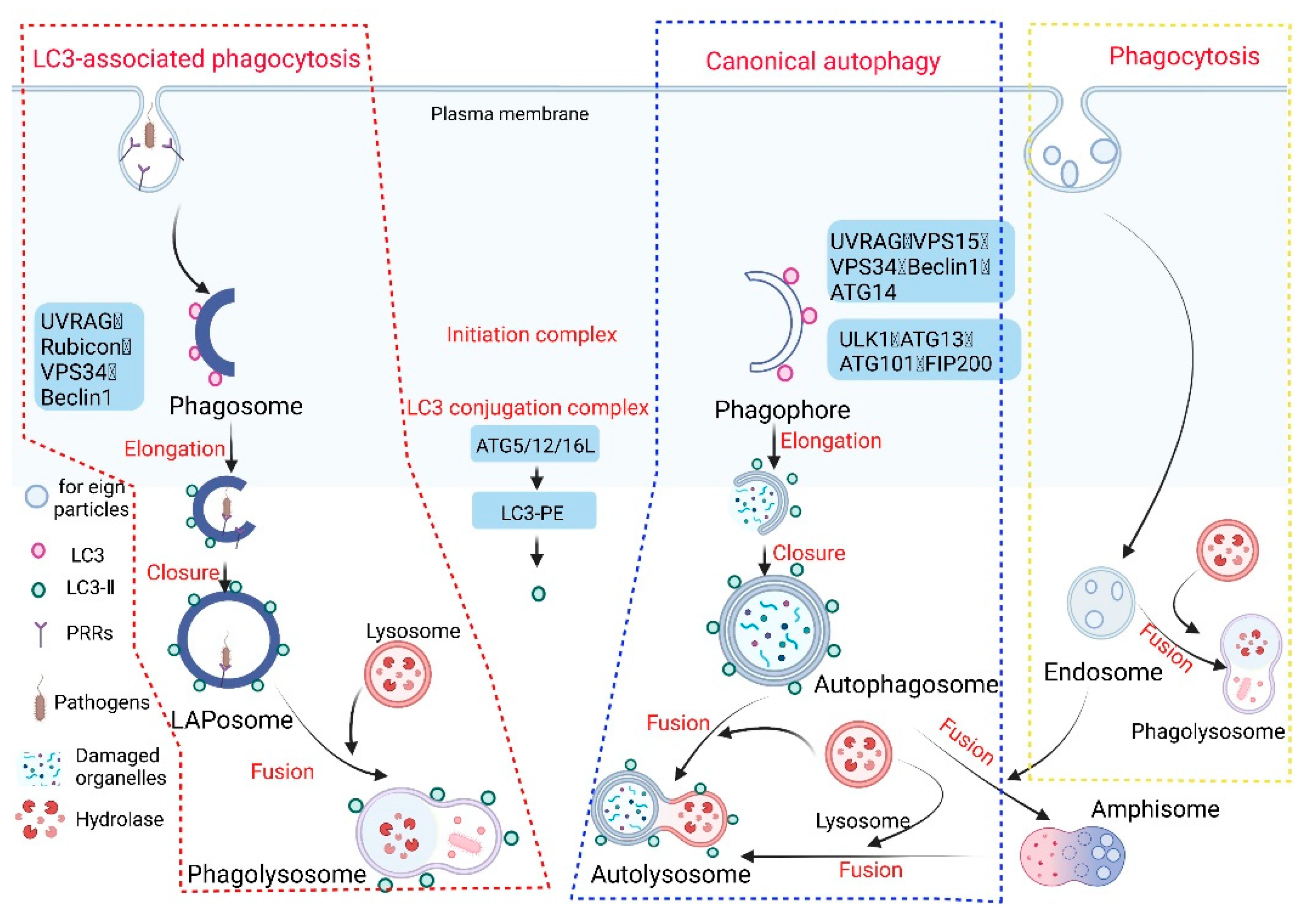
Figure 1. Phagocytosis, autophagy, and LC3-associated phagocytosis. Given the different origins of phagosome and autophagosome membranes, there are differences in the molecular mechanisms of LAP and autophagy. When foreign substances (e.g., apoptotic cells and invading pathogens) invade the host cell, specific receptors (such as Toll-like receptors, Fcγ receptors, or dectin-1) on the surface of the host phagocytes are activated to initiate LAP. The phagosome enables invading pathogens to be phagocytosed by the plasma membrane to form a single-membrane structure. Intracellular macromolecular proteins and damaged organelles are mainly surrounded by a double-membrane phagophore through autophagy, and then the phagosome and phagophore gradually extend and wrap around part of the cytoplasm and the organelles and proteins that need to be degraded in the cell to form the autophagosome and LAPosome. The autophagosome and endosome form an autophagosome (amphisome), and finally fuse with the lysosome to form the autophagolysosome and phagolysosome, respectively. A series of acid hydrolases are involved in the degradation of cytoplasmic substances to achieve cell homeostasis and organelle renewal (the red wireframe content represents the process of LAP occurrence, the blue wireframe content is the autophagy process, and the yellow wireframe content is the phagocytosis process).
Phagocytosis refers to the act of phagocytic cells ingesting solid particles from the surrounding environment in the form of protruding pseudopodia wraps. It is generally believed that the mechanism of this effect is the same as that of pinocytosis by the invagination of the cell membrane for the uptake of fluid or small molecules [2]. When the solid matter is adsorbed on the cell membrane, the membrane protrudes or sinks. Once the cell membranes on both sides are fused, the solid matter surrounded by the membrane is encapsulated in the cell. Phagocytosis in the body’s immune system is generally completed by professional phagocytes, which include dendritic cells, neutrophils, macrophages, and eosinophils [3]. These professional phagocytes activate phagocytosis of mammalian immune cells through attachment to pathogen-associated molecular patterns (PAMPs), which can lead to NF-κB activation. Opsonins such as C3b and antibodies can act as attachment sites, thereby immobilizing on the surface of phagocytes, and then promoting the internalization (uptake) of phagosomes through actin and myosin contraction systems, thereby forming phagosomes, which in turn ingest substances. The phagosomes fuse with lysosomes to form phagolysosomes and cause phagosome degradation.
Autophagy (referring to macroautophagy) is a process in which cells degrade their own proteins, organelles, or other intracellular components, and realize the reuse of degradation products and the renewal of organelles. Autophagy is dependent on the formation, maturation, and subcellular relocation of autophagosomes, ultimately leading to the fusion of autophagosomes with lysosomes. Autophagy can be divided into six stages: (1) pre-initiation, (2) autophagy initiation, (3) membrane vesicle extension, (4) autophagosome formation, (5) lysosome fusion, and (6) degradation and reuse. The molecular mechanism of autophagy involving multiple conserved autophagy-related proteins (ATG) has been widely described [4]. When the cell receives an autophagy-inducing signal, a membrane structure-like “liposome” is formed in the cytoplasm, and then continuously expands, forming a double-membrane structure under the electron microscope, which is called a phagophore. The phagophore will continue to extend until the components in the cytoplasm (e.g., misfolded proteins, damaged mitochondria) are surrounded, and an autophagosome is formed [5]. In this process, two ubiquitin-like coupling pathways are required, and then the fusion of autophagosome and lysosome will form an “autophagolysosome” or autolysosome, at which time the “cargo” endocytosed by the autophagosome will be degraded [6].
LAP is a noncanonical autophagy process reported in recent years in which microtubule-associated protein 1 light chain 3-β (LC3) binds to the phagosomal membrane using part of the autophagy mechanism [7]. Compared with autophagy that uptakes the cytoplasmic cargo, LAP is reported to target extracellular cargo. When pathogens invade the body initially, receptors on the surface of phagocytes, such as Toll-like receptors (TLRs), dendritic cell-associated C-type lectin-1 (dectin-1), and Fcγ receptors (FcγRs), recognize and interact with the PAMPs of pathogens to activate the LAP pathway [8][9][10][11][12]. The recruitment and assembly of the NADPH oxidase 2 (NOX2) complex on the initial phagosomes after LAP activation [13] is caused by the signaling cascade reaction of spleen tyrosine kinase (Syk) and protein kinase C (PKC), and is stabilized by the Rubicon protein. As part of the autophagy complex containing Beclin-1-vps34, Rubicon is a Beclin1-interacting protein and is essential for LAP maturation [14]. The recruitment of NADPH oxidase triggers the production of reactive oxygen species (ROS). ROS production leads to the rapid lipidation of LC3 and conjugation to the single-membrane phagosome which is the most different from a double-membrane vesicle in autophagy, thereby forming a vesicle decorated by LC3, which is called a LAPosome. Fusion of the LAPosomes with lysosomes results in their maturation into phagolysosomes, which can effectively eliminate engulfed pathogens, thereby improving the efficiency of phagocytosis and the killing of pathogens by phagolysosome complexes [15][16][17] (Figure 2).
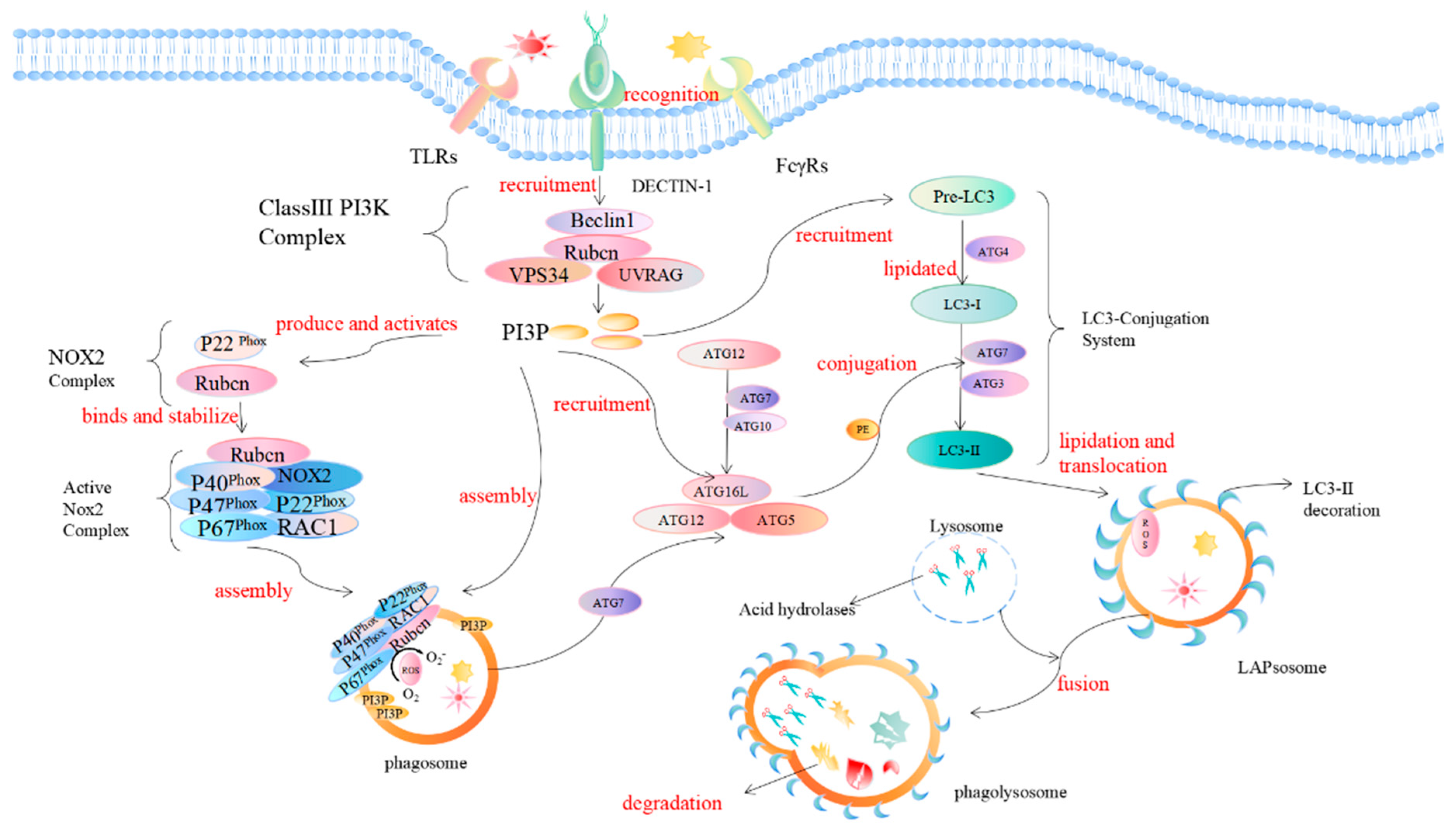
Figure 2. Mechanism of LC3-associated phagocytosis. During pathogen invasion, phagocyte surface-specific receptors (such as TLRs, FcγR, dectin-1) interact with PAMPs and activate the LAP pathway. Rubicon is recruited during LAP and promotes the activity of UVRAG-containing class III PI3K complexes. The PI3K complex, which is the first protein involved in LAP regulation, consists of Beclin-1, VPS34, UVRAG, and Rubicon. Rubicon maintains the stability of the PI3K complex, which in turn sustains the production of phosphatidylinositol 3-phosphate (PI3P) on the phagosome membrane, and PI3P acts as a downstream recruitment signal for autophagy. This process is necessary to stabilize the NOX2 complex, thereby maintaining the production of reactive oxygen species. PI3P is also critical for the complementation of components (such as ATG5, ATG3, ATG12, ATG7, and ATG16L) of the two subsequent ubiquitin-like conjugation systems (ATG5-ATG12-ATG16L, and the LC3-PE conjugation system) and the stabilization of the NOX2 complex to generate reactive oxygen species. The activation of the NADPH oxidase complex triggers the production of ROS on the phagosome. The production of ROS will lead to the rapid lipidation of LC3 in the phagosome membrane to form the LC3-associated phagosome (LAPosome), which is fused with the lysosome, and a series of acid hydrolases will participate in the degradation of cytoplasmic substances.
LAP is a unique pathway that links signaling during phagocytosis to the recruitment of certain components of the autophagic machinery. The regulation mechanism of autophagosome formation is an important part of autophagy research. LAP and autophagy are similar, but different. In the initiation process, autophagy begins with the formation of a double-membrane vesicle (phagophore) in the cytoplasm. In this process, two protein complexes are involved in autophagy: one containing the VPS34 complex (UVRAG, Beclin1, ATG14, VPS34, VPS15) and one containing the serine/threonine kinase ULK1 complex (ULK1, FIP200, ATG13, ATG101). LAP, on the other hand, involves the formation of a single-membrane phagosome, which involves initiation complexes including UVRAG, Beclin1, and VPS34, as well as a unique regulator, Rubicon. Among them, the Rubicon molecule acts like a switch, and although Rubicon inhibits VPS34 activity during autophagy, it is required for VPS34 activity on LAPosomes [18].
NADPH oxidase 2 (NOX2/gp91phox) was first discovered in phagocytes. NADPH oxidase consists of the common integral membrane protein subunit p22phox, the catalytic subunit gp91phox, the regulatory subunits p47phox, p40phox, and p67phox, and the small GTPase Rac. The gp91phox and p22phox subunits are mainly located on the plasma membrane [19]. The C-terminus of p22phox has a proline-rich region. The production of ROS requires the transfer of several other subunits (p47phox, p40phox, p67phox, and Rac) in the cytoplasm to the plasma membrane, following binding to gp91phox and p22phox. During LAP, Rubicon directly interacts with the p22phox subunit of NOX2 through its serine-rich region (amino acids 567–625) to stabilize the NOX2 complex for optimal and sustained ROS production, in which the NOX2 subunit p40phox binding to PI3P is also the result of the action of Rubicon [20]. Therefore, the stabilization of NOX2 and the generation of ROS are very important for LC3 lipidation during the subsequent LAP process and are also required for LAP formation [21]. LC3 plays a key role in the elongation and maturation of the autophagosome membrane. LC3 lipidation requires two ubiquitin-like conjugation systems, including the ATG5-ATG12 conjugation system and the LC3-phosphatidylethanolamine (PE) conjugation system on the surface of the phagosome. The first ubiquitin-like conjugation system activates ATG12 for ATG7, which can be linked to ATG10. ATG12 binds to ATG5 and further binds to ATG16L1 to form a multimeric ATG5–ATG12–ATG16L1 complex. The second ubiquitin-like conjugation system is the cleavage of cytoplasmic LC3 by ATG4 to generate LC3-I, which is activated by ATG7, and then covalently linked to PE on the membrane surface to convert LC3-I to lipidated LC3-II. The processed LC3-II is a marker for phagophores and phagosomes to mature into autophagosomes and LAPosomes, respectively. It is also a necessary condition for the autophagosome and LAPosome to fuse with the lysosome and degrade pathogens after fusion with the lysosome. While both autophagy and LAP are characterized by LC3-II binding to membranes, LC3-II is recruited to different types of membranes, which is one of the most striking ultrastructural differences that distinguish LAPosomes from autophagosomes.
2. Biological Functions of LAP
During bacterial infection, host cells engulf invading bacteria through a single-membrane vesicle called a “phagosome”. LC3 is recruited to the bacteria-containing phagosomal membrane to form an LC3-modified vesicle called a LAPosome. The LAPosome fuses with lysosomes to degrade bacteria. LAP begins with phagocytosis, transports captured pathogens to lysosomes for degradation, and enhances pathogen-killing efficiency through the LAPosome. LAP enhances innate immune cells’ ability to kill bacteria. LAP can transport bacteria in phagocytic vesicles to transmembrane pattern recognition receptors (PRRs) oriented toward the phagosomal lumen or cytosolic PRRs located within the host-cell cytosol for more efficient recognition and killing [22]. In addition, LAP promotes phagosome–lysosome fusion, enhances the degradation of bacteria, and plays an immunomodulatory role in antimicrobial immunity [23][24][25]. To survive in host cells, bacteria have also evolved strategies to evade targeted killing and degradation by LAP.
3. LAP and Bacterial Infection
The LAPosome fuses with lysosomes to degrade bacteria, but bacteria have evolved survival mechanisms to prevent this fusion process. The main mechanisms include blocking or destroying the production of phagosomes and impairing the fusion of phagosomes with lysosomes. Each of these two main methods creates an ideal environment for bacteria to replicate and survive in the body. Different bacteria have evolved various strategies to deal with LAP-promoted killing. Several typical strategies of bacteria in dealing with LAP are discussed below (Figure 3, Table 1).

Figure 3. Interactions of bacteria with LAP. This figure shows the correlation between several bacteria and LAP (black arrows). Green arrows indicate inhibition. Dotted arrows represent interactions that are not yet confirmed. L. monocytogenes uses its own virulence factors, PLCA/B and ActA, to protect it from being recognized, killed, and degraded by autophagy targets. PINCA did not play a substantial role in anti-L. monocytogenes and did not inhibit bacterial growth. L. monocytogenes promotes the uptake of mtCa2+ by regulating MCU to enhance PDH activity, thereby inducing the production of acetyl-CoA. Acetyl-CoA acetylates Rubicon, resulting in decreased Rubicon content and inhibiting the interaction of Rubicon with the NOX2 complex, thereby inhibiting the formation of LAP. The interaction of L. monocytogenes with Mac-1 induces ASMase-mediated changes in membrane lipid composition and converts sphingomyelin to ceramide and phosphorylated choline. The deletion of the PhoP and PurA virulence factors in S. typhimurium increased and decreased LC3 recruitment, respectively, and SsrB is part of the bacterial regulatory system that controls the expression of SPI2 effector molecules and is required for the maintenance of Salmonella-containing vacuoles (SCV). However, Rubicon knockout did not affect the survival of mutant SsrB strains, possibly because SPI2 is related to bacterial replication in vivo. CpsA acts upstream of NOX2 by blocking the recruitment of NOX2 to the phagosome; M. tuberculosis also secretes a virulence factor called NdkA. The presence of NdkA reduces the uptake of p67phox and Rac1 to the phagosome and interferes with NADPH oxidation. The enzyme complex generates ROS and may destroy LAP. LAP binds LC3 to LdCVs in a Dot/Icm T4SS-dependent manner, leading to bacterial degradation, a process that requires TLR2. L. pneumophila can irreversibly uncouple the conjugation function of LC3 and phosphatidylethanolamine through RavZ, and can also block LAP induced by its infection. BopA, an important effector protein encoded by the TTSS3 gene of B. pseudomallei, inhibits LC3 recruitment. The high and low expression of VAMP3 in Y. pseudotuberculosis can localize in autophagic vesicles of monolayer and bilayer membranes, respectively. VAMP7 inhibits the maturation of YCVs and disrupts LAP and autophagy, but the mechanism remains unclear. The spread of S. flexneri is dependent on the function of T3SS. IcsB blocks the recruitment of LC3 by blocking the binding of the ATG5 to the surface protein VirG of S. flexneri. IcsB is also thought to recruit the actin-associated protein TOCA-1, promoting cell-to-cell spread. Likewise, VirA prevents LC3 recruitment to S. flexneri-containing vesicles. Specifically, the inactivation of Rab1 by VirA inhibits the effect of ER on GA transport, but it remains unclear how Rab1 is involved in LC3 recruitment to S. flexneri-containing vacuoles. S. aureus was found to establish an intracellular niche in neutrophils, the mechanism of which has not been elucidated. S. pneumoniae-induced LC3 recruitment is dependent on pneumolysin.
Table 1. Bacterial pathogens and their evasion strategies.
| Bacteria | Virulence Factor | LAP-Specific Evasion Mechanisms | References |
|---|---|---|---|
| Legionella dumoffii | RavZ | Evasion of LAP by the T4SS effector protein RavZ, which inhibits LC3 lipidation and phagosome-lysosome fusion | [21][26] |
| Legionella pneumophila | RavZ | Evasion of LAP by the T4SS effector protein RavZ, which inhibits LC3 lipidation and phagosome–lysosome fusion | [26][27] |
| Burkholderia pseudomallei | BopA, BipD | Escapes from LAPosome and inhibits the recruitment of LC3 via BopA and BipD | [28][29][30] |
| Listeria monocytogenes | LLO | Upregulation of mitochondrial calcium signaling leads to the acetylation of Rubicon, interfering with LAP formation and the recruitment of NADPH oxidase | [23][24][31][32][33][34][35] |
| Streptococcus pneumoniae | PLY | Unknown | [36][37][38][39][40] |
| Mycobacterium tuberculosis | CpsA | Evasion of LAP by secreting CpsA protein to prevent NOX2 from assembling on phagosomes and inhibiting ROS production | [24][41][42][43][44][45][46] |
| Mycobacterium marinum | Type VII secretion systemESX1, CpsA | Evasion of LAP via non-acidifying LC3-positive vesicle, which is established through the ESX-1 secretion system | [47][48][49][50][51] |
| Shigella flexneri | IcsB, VirA | Evasion of LAPosome by inhibition of LC3 recruitment through interaction between TOCA-1 and IcsB | [52][53][54] |
| Yersinia pseudotuberculosis | Unknown | VAMP3 and VAMP7 co-localize with YCVs resulted in inhibiting LC3 recruitment | [55][56][57] |
| Salmonella enterica Typhimurium | PhoP, PurA, FlhD | Reduce TLR activation, inhibit LC3 recruitment, and inhibit phagolysosomal fusion | [58][59][60][61] |
| Staphylococcus aureus | Unknown | Forming spacious GFP-LC3-positive vacuoles that do not acidify | [45][62][63][64] |
References
- Huang, J.; Brumell, J.H. Autophagy in Immunity Against Intracellular Bacteria. Curr. Top. Microbiol. Immunol. 2009, 335, 189–215.
- Sosale, N.G.; Spinler, K.R.; Alvey, C.; Discher, D.E. Macrophage engulfment of a cell or nanoparticle is regulated by unavoidable opsonization, a species-specific ‘Marker of Self’ CD47, and target physical properties. Curr. Opin. Immunol. 2015, 35, 107–112.
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the Dead: Apoptotic Cell Sensing, Recognition, Engulfment, and Digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748.
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215.
- Pao, K.C.; Rape, M. Tug of War in the Xenophagy World. Trends Cell Biol. 2019, 29, 767–769.
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475.
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401.
- Pauwels, A.-M.; Trost, M.; Beyaert, R.; Hoffmann, E. Patterns, Receptors, and Signals: Regulation of Phagosome Maturation. Trends Immunol. 2017, 38, 407–422.
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257.
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical Autophagy Is Required for Type I Interferon Secretion in Response to DNA-Immune Complexes. Immunity 2012, 37, 986–997.
- Lamprinaki, D.; Beasy, G.; Zhekova, A.; Wittmann, A.; James, S.; Dicks, J.; Iwakura, Y.; Saijo, S.; Wang, X.; Chow, C.-W.; et al. LC3-Associated Phagocytosis Is Required for Dendritic Cell Inflammatory Cytokine Response to Gut Commensal Yeast Saccharomyces cerevisiae. Front. Immunol. 2017, 8, 1397.
- Hayashi, K.; Taura, M.; Iwasaki, A. The interaction between IKKα and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci. Signal. 2018, 11, eaan4144.
- Shahnazari, S.; Yen, W.-L.; Birmingham, C.L.; Shiu, J.; Namolovan, A.; Zheng, Y.T.; Nakayama, K.; Klionsky, D.J.; Brumell, J.H. A Diacylglycerol-Dependent Signaling Pathway Contributes to Regulation of Antibacterial Autophagy. Cell Host Microbe 2010, 8, 137–146.
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396.
- Underhill, D.M.; Rossnagle, E.; Lowell, C.A.; Simmons, R.M. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005, 106, 2543–2550.
- Tam, J.M.; Mansour, M.K.; Khan, N.S.; Seward, M.; Puranam, S.; Tanne, A.; Sokolovska, A.; Becker, C.E.; Acharya, M.; Baird, M.A.; et al. Dectin-1–Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. J. Infect. Dis. 2014, 210, 1844–1854.
- Herb, M.; Gluschko, A.; Schramm, M. LC3-associated phagocytosis—The highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 2020, 101, 68–76.
- Zhong, Y.; Wang, Q.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476.
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Yang, C.-S.; Lee, J.-S.; Rodgers, M.; Min, C.-K.; Lee, J.-Y.; Kim, H.J.; Lee, K.-H.; Kim, C.-J.; Oh, B.; Zandi, E.; et al. Autophagy Protein Rubicon Mediates Phagocytic NADPH Oxidase Activation in Response to Microbial Infection or TLR Stimulation. Cell Host Microbe 2012, 11, 264–276.
- Martinez, J.; Malireddi, R.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906.
- Ragland, S.A.; Kagan, J.C. Cytosolic detection of phagosomal bacteria—Mechanisms underlying PAMP exodus from the phagosome into the cytosol. Mol. Microbiol. 2021, 116, 1420–1432.
- Gluschko, A.; Herb, M.; Wiegmann, K.; Krut, O.; Neiss, W.F.; Utermöhlen, O.; Krönke, M.; Schramm, M. The β2 Integrin Mac-1 Induces Protective LC3-Associated Phagocytosis of Listeria monocytogenes. Cell Host Microbe 2018, 23, 324–337.e5.
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720.
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 2017, 429, 3561–3576.
- Hubber, A.; Kubori, T.; Coban, C.; Matsuzawa, T.; Ogawa, M.; Kawabata, T.; Yoshimori, T.; Nagai, H. Bacterial secretion system skews the fate of Legionella-containing vacuoles towards LC3-associated phagocytosis. Sci. Rep. 2017, 7, 44795.
- Rolando, M.; Escoll, P.; Nora, T.; Botti, J.; Boitez, V.; Bedia, C.; Daniels, C.; Abraham, G.; Stogios, P.J.; Skarina, T.; et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 1901–1906.
- Broek, C.W.V.; Stevens, J.M. Type III Secretion in the Melioidosis Pathogen Burkholderia pseudomallei. Front. Cell. Infect. Microbiol. 2017, 7, 255.
- Utaisincharoen, P.; Arjcharoen, S.; Lengwehasatit, I.; Limposuwan, K.; Sirisinha, S. Burkholderia pseudomallei invasion and activation of epithelial cells requires activation of p38 mitogen-activated protein kinase. Microb. Pathog. 2005, 38, 107–112.
- Gong, L.; Cullinane, M.; Treerat, P.; Ramm, G.; Prescott, M.; Adler, B.; Boyce, J.D.; Devenish, R.J. The Burkholderia pseudomallei Type III Secretion System and BopA Are Required for Evasion of LC3-Associated Phagocytosis. PLoS ONE 2011, 6, e17852.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345.
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578.
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192.
- Lam, G.Y.; Cemma, M.; Muise, A.M.; Higgins, D.E.; Brumell, J.H. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenesduring the early stages of macrophage infection. Autophagy 2013, 9, 985–995.
- Lam, G.Y.; Fattouh, R.; Muise, A.M.; Grinstein, S.; Higgins, D.E.; Brumell, J.H. Listeriolysin O Suppresses Phospholipase C-Mediated Activation of the Microbicidal NADPH Oxidase to Promote Listeria monocytogenes Infection. Cell Host Microbe 2011, 10, 627–634.
- Inomata, M.; Xu, S.; Chandra, P.; Meydani, S.N.; Takemura, G.; Philips, J.A.; Leong, J.M. Macrophage LC3-associated phagocytosis is an immune defense against Streptococcus pneumoniae that diminishes with host aging. Proc. Natl. Acad. Sci. USA 2020, 117, 33561–33569.
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659.
- Li, P.; Shi, J.; He, Q.; Hu, Q.; Wang, Y.Y.; Zhang, L.J.; Chan, W.T.; Chen, W.-X. Streptococcus pneumoniae Induces Autophagy through the Inhibition of the PI3K-I/Akt/mTOR Pathway and ROS Hypergeneration in A549 Cells. PLoS ONE 2015, 10, e0122753.
- Ogawa, M.; Matsuda, R.; Takada, N.; Tomokiyo, M.; Yamamoto, S.; Shizukuishi, S.; Yamaji, T.; Yoshikawa, Y.; Yoshida, M.; Tanida, I.; et al. Molecular mechanisms of Streptococcus pneumoniae -targeted autophagy via pneumolysin, Golgi-resident Rab41, and Nedd4-1-mediated K63-linked ubiquitination. Cell. Microbiol. 2018, 20, e12846.
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLOS Pathog. 2015, 11, e1005337.
- Romagnoli, A.; Etna, M.P.; Giacomini, E.; Pardini, M.; Remoli, M.E.; Corazzari, M.; Falasca, L.; Goletti, D.; Gafa, V.; Simeone, R.; et al. ESX-1 dependent impairment of autophagic flux byMycobacterium tuberculosisin human dendritic cells. Autophagy 2012, 8, 1357–1370.
- Ouimet, M.; Koster, S.; Sakowski, E.; Ramkhelawon, B.; Van Solingen, C.; Oldebeken, S.; Karunakaran, D.; Portal-Celhay, C.; Sheedy, F.J.; Ray, T.D.; et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 2016, 17, 677–686.
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreño, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 1–12.
- Shin, D.-M.; Jeon, B.-Y.; Lee, H.-M.; Jin, H.S.; Yuk, J.-M.; Song, C.-H.; Lee, S.-H.; Lee, Z.-W.; Cho, S.-N.; Kim, J.M.; et al. Mycobacterium tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death through Redox-dependent Signaling. PLoS Pathog. 2010, 6, e1001230.
- Van der Vaart, M.; Korbee, C.J.; Lamers, G.E.; Tengeler, A.C.; Hosseini, R.; Haks, M.C.; Ottenhoff, T.H.; Spaink, H.P.; Meijer, A.H. The DNA Damage-Regulated Autophagy Modulator DRAM1 Links Mycobacterial Recognition via TLR-MYD88 to Autophagic Defense. Cell Host Microbe 2014, 15, 753–767.
- Köster, S.; Klevorn, T.; Papavinasasundaram, K.; Sassetti, C.M.; Portal-Celhay, C.; Philips, J.A. Consequence of enhanced LC3-trafficking for a live, attenuated M. tuberculosis vaccine. Vaccine 2018, 36, 939–944.
- Lerena, M.C.; Colombo, M.I. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. 2011, 13, 814–835.
- Muñoz-Sánchez, S.; Van Der Vaart, M.; Meijer, A.H. Autophagy and Lc3-Associated Phagocytosis in Zebrafish Models of Bacterial Infections. Cells 2020, 9, 2372.
- Torraca, V.; Masud, S.; Spaink, H.P.; Meijer, A.H. Macrophage-pathogen interactions in infectious diseases: New therapeutic insights from the zebrafish host model. Dis. Model. Mech. 2014, 7, 785–797.
- Meijer, A.H. Protection and pathology in TB: Learning from the zebrafish model. Semin. Immunopathol. 2016, 38, 261–273.
- Wang, Q.; Zhu, L.; Jones, V.; Wang, C.; Hua, Y.; Shi, X.; Feng, X.; Jackson, M.; Niu, C.; Gao, Q. CpsA, a LytR-CpsA-Psr Family Protein in Mycobacterium marinum, Is Required for Cell Wall Integrity and Virulence. Infect. Immun. 2015, 83, 2844–2854.
- Weddle, E.; Agaisse, H. Spatial, Temporal, and Functional Assessment of LC3-Dependent Autophagy in Shigella flexneri Dissemination. Infect. Immun. 2018, 86, e00134-18.
- Campbell-Valois, F.-X.; Sachse, M.; Sansonetti, P.J.; Parsot, C. Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. mBio 2015, 6, e02567-14.
- De Lichtenberg, U.; Jensen, L.J.; Fausbøll, A.; Jensen, T.S.; Bork, P.; Brunak, S. Comparison of computational methods for the identification of cell cycle-regulated genes. Bioinformatics 2005, 21, 1164–1171.
- Lemarignier, M.; Pizarro-Cerdá, J. Autophagy and Intracellular Membrane Trafficking Subversion by Pathogenic Yersinia Species. Biomolecules 2020, 10, 1637.
- Ligeon, L.-A.; Moreau, K.; Barois, N.; Bongiovanni, A.; Lacorre, D.-A.; Werkmeister, E.; Proux-Gillardeaux, V.; Galli, T.; Lafont, F. Role of VAMP3 and VAMP7 in the commitment ofYersinia pseudotuberculosisto LC3-associated pathways involving single- or double-membrane vacuoles. Autophagy 2014, 10, 1588–1602.
- Moreau, K.; Lacas-Gervais, S.; Fujita, N.; Sebbane, F.; Yoshimori, T.; Simonet, M.; Lafont, F. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell. Microbiol. 2010, 12, 1108–1123.
- Masud, S.; van der Burg, L.; Storm, L.; Prajsnar, T.K.; Meijer, A.H. Rubicon-Dependent Lc3 Recruitment to Salmonella-Containing Phagosomes Is a Host Defense Mechanism Triggered Independently From Major Bacterial Virulence Factors. Front. Cell. Infect. Microbiol. 2019, 9, 279.
- Wu, S.-Y.; Wang, L.-D.; Li, J.-L.; Xu, G.-M.; He, M.-L.; Li, Y.-Y.; Huang, R. Salmonella spv locus suppresses host innate immune responses to bacterial infection. Fish Shellfish Immunol. 2016, 58, 387–396.
- Wang, H.; Liu, Y.; Ye, D.; Li, J.; Liu, J.; Deng, F. Knockdown of zebrafish Nanog increases primordial germ cells during early embryonic development. Dev. Growth Differ. 2016, 58, 355–366.
- Wang, L.; Li, Y.; Liu, Y.; Zuo, L.; Li, Y.; Wu, S.; Huang, R. Salmonella spv locus affects type I interferon response and the chemotaxis of neutrophils via suppressing autophagy. Fish Shellfish Immunol. 2019, 87, 721–729.
- Masud, S.; Prajsnar, T.K.; Torraca, V.; Lamers, G.E.M.; Benning, M.; Van Der Vaart, M.; Meijer, A.H. Macrophages target Salmonella by Lc3-associated phagocytosis in a systemic infection model. Autophagy 2019, 15, 796–812.
- Prajsnar, T.K.; Serba, J.J.; Dekker, B.M.; Gibson, J.F.; Masud, S.; Fleming, A.; Johnston, S.A.; Renshaw, S.A.; Meijer, A.H. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy 2021, 17, 888–902.
- Pollitt, E.J.G.; Szkuta, P.T.; Burns, N.; Foster, S.J. Staphylococcus aureus infection dynamics. PLoS Pathog. 2018, 14, e1007112.
More
Information
Subjects:
Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
15 Aug 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No