Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hasi Rani Barai | -- | 5092 | 2022-08-06 02:44:32 | | | |
| 2 | Rita Xu | -3 word(s) | 5089 | 2022-08-08 04:46:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pardeshi, S.; Damiri, F.; Zehravi, M.; Joshi, R.; Kapare, H.; Prajapati, M.K.; Munot, N.; Berrada, M.; Giram, P.S.; Rojekar, S.; et al. Thermoresponsive Hydrogel Molecule. Encyclopedia. Available online: https://encyclopedia.pub/entry/25914 (accessed on 22 July 2026).
Pardeshi S, Damiri F, Zehravi M, Joshi R, Kapare H, Prajapati MK, et al. Thermoresponsive Hydrogel Molecule. Encyclopedia. Available at: https://encyclopedia.pub/entry/25914. Accessed July 22, 2026.
Pardeshi, Sagar, Fouad Damiri, Mehrukh Zehravi, Rohit Joshi, Harshad Kapare, Mahendra Kumar Prajapati, Neha Munot, Mohammed Berrada, Prabhanjan S. Giram, Satish Rojekar, et al. "Thermoresponsive Hydrogel Molecule" Encyclopedia, https://encyclopedia.pub/entry/25914 (accessed July 22, 2026).
Pardeshi, S., Damiri, F., Zehravi, M., Joshi, R., Kapare, H., Prajapati, M.K., Munot, N., Berrada, M., Giram, P.S., Rojekar, S., Ali, F., Rahman, M.H., & Barai, H.R. (2022, August 06). Thermoresponsive Hydrogel Molecule. In Encyclopedia. https://encyclopedia.pub/entry/25914
Pardeshi, Sagar, et al. "Thermoresponsive Hydrogel Molecule." Encyclopedia. Web. 06 August, 2022.
Copy Citation
Temperature-induced, rapid changes in the viscosity and reproducible 3-D structure formation makes thermos-sensitive hydrogels an ideal delivery system to act as a cell scaffold or a drug reservoir. Moreover, the hydrogels’ minimum invasiveness, high biocompatibility, and facile elimination from the body have gathered a lot of attention from researchers.
hydrogel
thermoresponsive
release kinetics
1. Introduction
A hydrogel is a three-dimensional macromolecule network that is interlinked. It comprises hydrophilic or amphiphilic building blocks, which swell in water and may hold a lot of it. Cross linking makes hydrogels insoluble in water. To form a gel, water must account for at least 10% of the total volume or weight. Micro-sized water-soluble monomers, nanosize nanofibrils or nanotubes, and polymers make up hydrogels [1][2]. Crosslinking is a crucial characteristic that inhibits the gel from dissolving in water. Chemical and physical crosslinking are the two forms of crosslinking [3]. There is a covalent bond formed in chemical crosslinking, similar to the development of disulfide linkage, and this kind of gel would be permanent. Intermolecular forces, such as hydrogen bonding and other hydrophobic interactions or physical entanglements, are engaged in physical crosslinking [4]. Hydrogels have become the focus of considerable research in recent years. Their features, such as high-water content and the ability to control swelling characteristics, make them particularly appealing for biomedical applications [5]. In situ synthesized hydrogels can be used to create easy, “custom-made” treatments and diagnostics. Following the polymerization process, chemical or ionic crosslinking, or in reaction to an environmental stimulus such as temperature, pH, or ionic concentration of the external medium, a polymeric solution can be created and allowed to gel in situ [6].
Similarly, nanogels are nanosized hydrogels that combine the benefits of nanoscale materials with the benefits of hydrogels. Nanogels are distinguished by their small dimensions (up to 1000 nm) with high water absorption capacity. Thermoresponsive nanogels are delicate nanomaterials that react to variations in temperature. To make thermoresponsive nanogels, two methods have been used [7]. The primary structure of a nanogel-forming polymer is integrated with thermoresponsive polymer units in the first strategy to create thermoresponsiveness. To provide thermoresponsiveness, lipophilic moieties are bonded as a substituent to a hydrophilic polymer backbone in the second technique. The amount of interaction, which can be classified as hydrophobic or hydrophilic based on the change in free energy of the surrounding solvent, is the factor that causes thermoresponsiveness [8]. The hydrophobicity or hydrophilicity of a combination is indicated by a positive or negative variation in its free energy. Tailored drug delivery, the modulated release of drugs, imaging and tracking, and other applications of thermosensitive nanogels have all been investigated. Peters et al. developed a thermoresponsive PNIPMAM-based core-shell nanogel for the treatment of cancer that allows for regulated and triggered DOX release. With L929 fibroblasts, the toxicity of the produced nanogels was studied, which revealed low inherent cytotoxicity [8][9].
The hydrogels that react to changes in temperature. Their responsiveness to the thermal sensation is advantageous because heat is the single trigger for their gel formation, requiring no further chemical or environmental intervention. It can therefore be created, for example, when the temperature rises from atmospheric to physiological. Sol-gel shift is the term that describes the transformation from a solution to a gel [3][6][10]. Beyond a certain temperature, some hydrogels separate from the solution and solidify, and this threshold is defined as the lower critical solution temperature (LCST). The polymers are solvable below the LCST. They become progressively hydrophobic and insoluble above the LCST, resulting in gelation. Hydrogels generated by cooling a polymer solution, on the other hand, have an upper critical solution temperature (UCST). Spectroscopy, differential scanning calorimetry (DSC), and rheology can be used to confirm the sol-gel transformation of thermo-responsive hydrogels in the lab [11]. Due to their distinctive features, thermoresponsive hydrogels are extremely fascinating.
In the aqueous phase, poly(N-isopropylacrylamide) (NIPAAm) based polymers often had a very well defined lower critical solution temperature (LCST) of 32 °C, during which hydrogen bonds among water molecules and the NIPAAm chain form/break, accompanied by network hydration/dehydration. The swelling behavior of NIPAAm hydrogels varies dramatically over a small temperature range due to the delicate equilibrium between hydrophilic and hydrophobic nature [6][12]. Its temperature-modulated swelling feature has been used to create an on/off switch for various applications, including medication administration, controlled release, and so on. Introducing a hydrophobic or hydrophilic comonomer/crosslinker into the LCST could also be used to change the hydrophilic/hydrophobic equilibrium inside the polymer. Biomedical applications often demand the LCST between room temperature and body temperature (37 °C) and even higher [10][11].
2. Stimuli-Responsive Polymers
Temperature, ion concentration, redox processes, pH, light, shear stress, enzymes, and other triggers initiate stimuli-responsive polymers. The macroscopic behavior of polymer solutions is determined by their physical characteristics [13]. This even allows for some customization over the characteristics of biomedical devices and delivery systems comprised of such polymers, which has sparked a huge interest. The biological applicability of stimulus-responsive polymer systems allows only a limited range of stimuli, such as pH, ionic strength, and others, which limits their applicability. In this temperature range, various types of polymers exhibit thermosensitive characteristics [14].
2.1. Physicochemical Properties of Thermoresponsive Hydrogel
2.1.1. Mechanical Strength
Thermosensitive hydrogels are delicate materials that alter physical characteristics in response to temperature changes, and they have a lot of applications in biomedicine [15][16]. Existing thermoresponsive hydrogels, such as poly(N-isopropylacrylamide) (PNIPAM), have poor mechanical properties and lack an energy-dissipating mechanism, making them unsuitable for potential implementation. Although some techniques, such as double networks, dual-cross-linking, and composites, can increase strength, they can diminish thermoresponsiveness. The development of thermosensitive hydrogels that retain inherent mechanical characteristics remains an important and pressing challenge. Hydrogel actuators with high strength, hardness, and stimuli sensitivity can be made by molecularly combining a hard element and a stimuli-sensitive component in a single polymer network [17]. Hydrogels that combine thermosensitive and hard compounds into a single system have recently been introduced by scientists at the University of California, resulting in high resilience and thermal responsive performance [18]. Thermal responsive PNIPAM was anchored onto the tough polymer backbone of poly (vinyl alcohol) (PVA) and its methacrylate derivatives using UV light irradiation (PVA-MA). The samples were dipped in a sodium sulphate salt solution after the one-pot polymerization to further enhance the hydrogel network, in which the PVA clumps and crystallizes under these circumstances. This hardening process does not decrease the stimuli-responsive qualities due to the single-network topology. The new hydrogel performed well mechanically and had a toughness (~10 MJ/m3) 100 times that of ordinary PNIPAM hydrogels [19].
2.1.2. Adhesion
The development of robust and effective bonding among hydrogels and solid substances in wet environments, which would be critical for its assimilation into and efficiency in systems and devices, is still a continuous process. The excessive quantity of water in the polymeric matrix of hydrogels weakens their adhesive behaviour, as water generates a weak barrier that prevents actual surface contact between the hydrogels and substrates, resulting in reduced surface energy and decrease of adhesion strength [20]. Additionally, water molecules connect with the adhesive sites in the hydrogels via hydrogen bonding, reducing interfacial adhesion between the hydrogels and solid substances dramatically. If hydrogels are used in biomedicine, the situation becomes much more complex because most substrates (human bodies, tissues, and bioglues) are moist and delicate [15]. Researchers have investigated the chemical properties of marine animals, e.g., sandcastle worms and mytilus mussels, which can attach to a variety of immersed substrates in a hostile and unstable wet environment, to improve the adherence of a hydrogel to wet surfaces. Wet adhesives must meet numerous critical characteristics in comparison to dry adhesives, includes breaking down surface bound water layers, keeping cohesive against water corruption, and having a planned underwater healing [21]. Waite and Tanzer discovered a Catecholic amino acid (DOPA) in mussel foot plaque in 1981, and discovered that it contributes a lot to mussels’ powerful underwater adherence [22]. The catechol-containing peptide was found to be capable of penetrating aqueous boundary barriers and forming interfacial connections with underwater surfaces [17].
2.1.3. Optical Property
Sensing, optoelectronics, nanomotion, theranostics, and biomedical applications are just a few of the uses for plasmonic compounds [23]. Plasmonic particles covered in thermosensitive polymers have plasmonic signals that are heat sensitive, which is of special importance. Poly-N-isopropyl acrylamide is commonly used in these systems (pNIPAM). At relatively low temperature, the polymer chains of pNIPAM are lengthened, and once the lower critical solution temperature (LCST) is achieved, the polymer experiences a hydrophilic-to-hydrophobic shift and collapses in aqueous phase [24]. As a result, in a gel comprising pNIPAM and plasmonic nanomaterials, the plasmonic signature changes due to either (1) an alteration in pairing in between nanomaterials as a feature of their detachment, (2) a modification in the refractive index of the polymer underlying the nanomaterials due to the removal of water from the polymer and substitution of the water with a greater refractive index organic polymer, or (3) both pathways [25]. Yang and colleagues demonstrated how these heat-sensitive substances might be employed, wherein silver nanoparticles were produced in situ within a polymeric gel consisting of pNIPAM [26]. Thermal responsive surface-enhanced Raman scattering (SERS) responses arise from a variation in polymer structure with heat. Feldman and colleagues demonstrated that optoelectronic actuators may be made using microgels of pNIPAM-coated gold nanorods. Other heat-responsive polymers could be used, as demonstrated by Liz-Marzan and colleagues [27]. They have used oligo (ethylene oxide) methacrylate as a heat responsive polymer conjugated from gold nanoparticles altered with bovine serum albumin whereby an atom transfer radical polymerization initiator was covalently attached [28][29].
Lasting Time
The lasting time or residence time of thermoresponsive hydrogels depends on various parameters, such as the nature and structure of the monomer, degree of cross-linking of the polymer, rate of biodegradation of polymer, or the intensity of the thermal stimulus. It plays an important role in deciding the fate of drug delivery. Plain thermosensitive polymers have poor mechanical strength and thus may get degraded in the biological fluid, rapidly limiting the sustained release of entrapped drugs. These polymers can be coupled with polymers having high mechanical strengths. Thus, the formed copolymers will have a greater lasting time as their degradation rate will be lowered, hence possibly leading to a prolonged residence time of the drug, ultimately improving the efficacy. Cui Z et al. studied the degradation property of copolymers synthesized using NIPAAm. It was found that at physiological conditions, LCST increased to approximately 35 °C from 37 °C after 20 days, indicating a slower rate of degradation and increased lasting time. It was also dependent on the mass loss property of the copolymer. Md Hasan Turabee et al., Synthesized N, N, N-trimethyl chitosan embedded thermosensitive Pluronic F127 hydrogel for the treatment of brain tumor. It was seen that, at pH 7.4, even after 30 days, the synthesized gel sustained the release of docetaxel for the treatment of glioma [30][31].
2.2. Natural Polymers and Their Derivatives
Many natural polymers and their derivates form a gel with temperature change. Thermosensitive polymers vary their solubility in response to changes in ambient temperature. Thermally responsive hydrogels can be made using these polymers alone or in combination with synthetic polymers [32].
2.2.1. Polysaccharides
Cellulose Derivatives
Cellulose is insoluble in water. It was modified with methyl or hydroxypropyl groups to increase its water solubility. Methylcellulose (MC), a derivate of it, has been intensively studied for biomedical purposes. MC produces a thermoresponsive gel at 60–80 °C that cools to become a solution [33]. Methylcellulose was substituted with N-isopropyl acrylamide (NiPAAm), and the resulting polymer showed combined thermo-gelling characteristics of both polymers. Researchers also discovered that adding MC to NiPAAM polymers improves the hydrogel’s mechanical properties [34]. Stabenfeldt et al. functionalized methylcellulose with the laminin to fabricate a bioactive scaffold for neural tissue engineering. Methylcellulose was first oxidized (OXMC), followed by laminin tethering (OXMC+LN). OXMC+LN hydrogel enhanced neuronal cell adhesion and cell viability compared to MC and OXMC [35].
Chitosan
The deacetylation of chitin, which is found in the exoskeleton of crustaceans and insects, produces chitosan. Bhattarai et al. grafted poly (ethylene glycol) (PEG) into chitosan. PEG grafting improved the solubility of chitosan in water. Without a crosslinker, the PEG-grafted chitosan can form a thermoreversible hydrogel in physiological pH values [36].
Once evaluated with the model drug albumin, PEG-grafted chitosan exhibited controlled in-vitro drug release with an early burst and a continuous release for three days. When the PEG-grafted chitosan was crosslinked in situ using genipin, a low-cytotoxicity crosslinking agent, the quasi-linear release of drugs for up to 40 days was achievable. At 37 °C, though, the thermoreversible property of the hydrogel was lost [37]. A copolymer of chitosan of NiPAAm was prepared. The resultant hydrogel showed potential as cell carriers for tissue engineering applications and can be used to treat vesicoureteral reflux with minimal invasion [38]. Chitosan was conjugated with hydroxybutyl groups by Dang et al. [24]. At physiological temperatures, the formed hydrogel may gel in seconds and cool to a liquid state. When enclosed in hydrogels over two weeks, hMSC and cells originating from the intervertebral disc increased and produced an extracellular matrix. A chitosan-glycerophosphate salt (GP) hydrogel was tested for neural tissue engineering applications. Crompton et al. tested this hydrogel to polylysine-functionalized chitosan-GP to improve neuronal attachment and neurite development. More cells survived over nonmodified chitosan-GP at particular polylysine concentrations. This shows that polylysine-chitosan-GP could be a viable choice for brain tissue engineering [39].
Dextran
Dex-MA was created by crosslinking biodegradable dextran (Dex) with maleic anhydride (MA). Photo crosslinking the copolymer with NiPAAm made it more thermoresponsive. The resultant hydrogel was pH-sensitive and partly biodegradable [11]. Huang and colleagues used NiPAAm to copolymerize dextran oligolactate and 2-hydroxyethyl methacrylate (Dex-lactate-HEMA). The scientists determined that the drug release pattern is influenced by various parameters, including temperature, hydrogel swelling and degradation properties, and drug–hydrogel macromolecule interaction [40].
Xyloglucan
If more than 35% of the galactose remnants in xyloglucan are eliminated, it exhibits thermosensitive activity. Xyloglucan gels have been used to deliver drugs in a range of applications. Unfortunately, there is a lack of information on these hydrogels’ viscoelastic and morphological properties [41]. Nisbet et al. investigated the gelation characteristics and morphology of xyloglucan hydrogels under physiological circumstances. In comparison to deionized water, the existence of ions in PBS seems to affect the gelation. The optimum amount was discovered to be 3% (wt.) xyloglucan in aqueous media, which has a much greater elastic modulus than other natural or synthetic hydrogels. Furthermore, a gel might be freeze-dried at this concentration and analyzed using scanning electron microscopy. The images showed a macroporous, interconnected, three-dimensional network [42].
2.2.2. Proteins
Gelatin
Gelatin is a thermoresponsive, biocompatible, and biodegradable polymer. The gelatin aqueous solution solidifies under 25 °C, while the gel reverts to fluid beyond 30 °C. Gelatin coupled with the other polymers demonstrates thermoresponsive gel formation near body temperature, which is desirable for biomedical applications [43]. Yang et al. created a gelatin hydrogel with monomethoxy poly (ethylene glycol)-poly (D, L-lactide) (mPEG-DLLA) block copolymers. At 37 °C, gelatin was combined with 30% wt. mPEG-DLLA and gelled quickly. Using gentamycin sulphate, hydrogels take five days or longer to release 50% of medication at ambient temperature. The whole duration of the release is 40 days. At 37 °C, the release profile is much slower.
Nevertheless, due to the deterioration of the hydrogel matrices, the release was no longer measurable after a week [44]. Ohya and Matsuda created a thermosensitive gel by grafting gelatin with NiPAAm. The resultant polymer displays a sol-gel transformation at physiologic temperatures and a pNiPAAm/gelatin (P/G) ratio larger than 5.8. Due to improved hydrophobicity and a favorable cell environment, a moderate hydrogel concentration (5% w/v) and a high P/G ratio resulted in higher cell growth and extracellular matrix formation [45]. Gil et al. created a thermoresponsive gel by combining gelatin and silk fibroin [33]. Silk fibroin crystals stabilized the gel at 37 °C. The gel swelled more at physiological temperatures than at 20 °C, but it also lost more mass due to gelatin disintegration and was released [46].
2.2.3. Synthetic polymers and their derivatives
N-Isopropylacrylamide(pNiPAAm)-Based Systems
Thermoresponsive hydrogels based on pNiPAAm and its derivatives have been investigated extensively for drug delivery, cell encapsulation and cell culture surfaces. Coughlan and colleagues observed that with Hydrogel, the swelling was decreased in the presence of hydrophobic drugs, and the opposite effect was observed for hydrophilic drugs. The authors suggested that drug properties such as solubility, size, and chemical nature should be considered when choosing pNiPAAm hydrogel as a delivery vehicle [47]. The thermoreversible p(NiPAAm-co-AA) hydrogel was tested as a cell and drug delivery vehicle [48].
Liu et al. have polymerized p(NiPAAm-co-AA) with ethyl acrylate (EA). The swelling was lower due to the hydrophobic pEA. P(NiPAAm-coAA) showed an initial burst release, not observed on p(pNiPAAm-co-AA)/pEA. It was concluded that the pEA chains had a favorable effect on maintaining a slower and more stable release profile [49]. Yin et al. synthesized copolymers using NiPAAm and PAA. They showed that even small changes in pH could significantly affect the hydrogel thermo-responsiveness. This feature can be useful for applications such as drug delivery, where physiological temperature and local pH differences can both act as stimuli, and for molecular switching over the desired pH range [50]. Xu et al. formed a triblock copolymer hydrogel that showed a combination of stimuli-responsive attributes. A poly((2-dimethyl amino) ethyl methacrylate-co-2-hydroxyethyl methacrylate)-b-poly(N-isopropyl acrylamide)- b-poly((2-dimethyl amino) ethyl methacrylate-co-2-hydroxyethyl methacrylate) or p(DMAEMA-co-HEMA)-b-p(NiPAAm)-b-p(DMAEMA-co-HEMA) copolymer was synthesized by atom transfer radical polymerizations (ATPR). Its temperature-responsive behavior was attributed to pNiPAAm and the pH sensitivity to pDMAEMA [51]. A copolymer of pentaerythritol monostearate diacrylate (PEDAS), N-isopropyl acrylamide (NiPAAm), acrylamide (AAm), and 2-hydroxyethyl acrylate (HEA) was synthesized. PEDAS contains a lipophilic side chain, and AAm and HEA can modulate hydrophilicity and add groups for subsequent acrylation and crosslinking. The thermal gelation is attributed to the NiPAAm block. Future work is directed to be addressed biodegradability. Many homo-and copolymers of NiPAAm are not biodegradable, which may prove problematic for some biomedical engineering applications [52]. Nakayama and colleagues have prepared thermally responsive, biodegradable micelles that incorporate water-insoluble drugs. By combining a poly(N-isopropyl acrylamide-co-N, N-dimethyl acrylamide) (p(NiPAAm-co-DMAAm)) block, which has an LCST around 40 °C, with poly(D, L-lactide), poly(ε-caprolactone) or poly(D, L-lactide-co-ε-caprolactone), which are all biodegradable and hydrophobic, the group was able to fabricate polymeric micelles with controlled dimensions and phase transition temperatures [53]. Recently, Hatakeyama et al. have produced bioactive, thermoresponsive cell culture surfaces by immobilizing the cell adhesive peptide RGDS and the growth factor insulin on a NiPAAm-copolymer. N-Isopropylacrylamide was copolymerized with its analog 2-carboxy isopropyl acrylamide, and the polymer was grafted onto polystyrene tissue culture dishes, followed by RGDS and insulin immobilization. They found that these factors increase cell adhesion and proliferation, reducing culture time. When the temperature was brought to 20 °C, the cells could be quickly recovered as contiguous tissue monolayers [54].
2.2.4. PEO/PPO-Based Systems
Triblock copolymers poly (ethylene oxide)-b-poly (propylene oxide)-b-poly (ethylene oxide) (PEO-PPO-PEO), also known as Pluronics® or Poloxamers, exhibit a thermoreversible behavior at physiological temperature and pH [48]. The amphiphilic polymer structure has hydrophilic ethylene oxide and hydrophobic propylene oxide. The gelation occurs as changes in micellar properties function of both concentration and temperature. Above critical micelle concentration (CMC), the amphiphilic block copolymer molecules can self-assemble into micelles. Pluronics® polymer drug delivery exhibits a CMC of 1 μM to 1 mM at 37 °C. Polypropylene oxide is relatively soluble in the water below critical micelle temperature (CMT). With a temperature increase, polypropylene oxide chains become less soluble, resulting in micelle formation [55]. The commonly used Pluronics® in biomedical applications is F127 has a weight percentage of 70% PEO and a molecular weight of PPO around 4000 [56]. Pluronics have been extensively used in drug and gene delivery, inhibition of tissue adhesion, and burn wound covering. Pluronics® can be a suitable substrate for hematopoietic stem cells, supporting their culture and preservation and tissue engineering applications. F127 was also evaluated as a scaffold for lung tissue engineering, showing promising results on tissue growth with a low inflammatory response [57]. Wein and colleagues tested a β-tricalcium phosphate (β-TCP) scaffold, using an F127 hydrogel to facilitate cell delivery and distribution for an in vitro study aiming at bone regeneration. They reported that F127 was no longer present in the channels of the β-TCP scaffold after one week in culture and seemed to have degraded. Bone tissue growth was only weakly induced, and the constructs showed lower stiffness than other hydrogels (fibrin, collagen I) composites evaluated [58]. Cohn and colleagues proposed two new mechanisms to create copolymers based on Pluronic® F127 with improved mechanical properties. In both cases, they relied on the principle of a multiblock backbone with the addition of covalently bound repeating units. Both newly synthesized polymers exhibited significantly higher viscosities than F127 at 37 °C, and the poly(ether-urethanes) displayed much slower drug release kinetics than the original polymer [59]. Pluronic® polymers were functionalized with acrylic moieties and thiols at their end groups and were subsequently gelled at 37 °C by Cellesi et al. It was found that these polymers were biocompatible, allowing for the encapsulation of sensitive drugs and cells. A similar method was followed with Tetronic® polymers, which are thermosensitive, tetra-armed Pluronic® analogs. By adjusting the molecular weight of the precursors and the functionalization (therefore also the crosslinking density), the final mechanical and transport properties of the “tandem” polymers can be controlled. Moreover, the “tandem” method allows for easy processing of the polymers, e.g., into spherical beads and hollow capsules [60].
2.2.5. PEG/Biodegradable Polyester Copolymers
Thermoresponsive properties were ascertained by adjusting the hydrophobic polyester block and the PEG block length appropriately. These polymers were biocompatible, biodegradable, and exhibited a sol-gel transition. The use of high molecular weight-PLGA combined with low molecular weight-PEG resulted in a hydrogel with quick gelation at physiological temperature. The combination of hydrophobic/hydrophilic units created a surfactant behavior of the polymers in water, thus facilitating the solubilization of hydrophobic drugs. In vivo studies showed good mechanical properties and integrity for longer than a month [61]. More recently, Chen et al. developed a triblock PLGA-PEG-PLGA-based system for the controlled release of testosterone. Testosterone is water-insoluble, and so far, its delivery systems have included patches, creams, gels, injectables, and implants. A slower in vitro release of testosterone was observed for copolymers with longer PLGA blocks, possibly due to the slower degradation of these hydrophobic units. The thermosensitive polymers showed a controlled, linear release for three months [62]. Another recent approach towards a thermoresponsive system involved the synthesis of a multiblock copolymer with a biodegradable polyester. Alternating multiblock poly (ethylene glycol)/poly(L-lactic acid) (PEG/PLLA) copolymers were produced. It was shown that sol-to-gel transition depends on the total molecular weight (MW) and the MW of each building block. In vitro and in vivo gelation studies determined that a copolymer with a total MW of 6700 daltons and 600/1300 (MW of PEG/PLLA blocks, respectively) holds potential as an injectable carrier for biomedical applications in terms of transition temperature and modulus at 37 °C [63].
2.2.6. Poly(organophosphazenes)
Current advances in poly(organophosphazenes) include their use as a drug [64] and cell delivery systems. Poly(organophosphazenes) grafted with mPEG and amino acid esters were reported as a new class of biodegradable and thermosensitive polymers in 1999 [52]. Sohn and colleagues developed a correlation for the LCST of these polymers as a function of their molecular structure, which comprises hydrophilic (PEG) and hydrophobic (amino acid esters) side groups. Polymers showed a sustained release profile for both hydrophobic and hydrophilic drugs. Moreover, their use as cell carriers holds promise. More recently, hydrogels exhibiting a thermosensitive sol-gel behavior have been reported as cell carriers for tissue regeneration. Typically, aqueous solutions of hydrogels used in biomedical applications are liquid at ambient temperature and gel at 37 °C [34][65].
3. Mechanism of Thermoresponsive Hydrogel
Thermoresponsive hydrogels originate from phase transition from gel phase to solution-phase and vice versa with temperature variation. The interaction of surrounding phase and functional group of polymers important for gel formation. In the case of a thermoresponsive hydrogel, the cross-linking ability has achieved widespread biomedical application. Poly(N-isopropyl acrylamide) (PNIPAM) is an ideal natural thermoresponsive polymer [66]. The temperature at which a functional copolymer undergoes the transition from solution phase to gel phase is called a sol-gel phase transition. One class of functional polymeric material transformed to a solid phase (gel-like consistency) and separated from the solution above a specific temperature threshold limit, defined as the lower critical solution temperature (LCST). However, another class of functional polymer exhibited a solid phase (gel-like consistency) after cooling their solutions. Another threshold limit is upper critical solution temperature (UCST) [65][67].
Thermoresponsive polymers which solubilize in an aqueous solvent or organic solvents are not suitable for biomedical applications. As shown in Figure 1. LCST polymers are soluble below a critical temperature, whereas UCST polymers solubilize above UCST. The water in thermo-responsive hydrogels is altered by small changes in temperature and the resulting polymeric chain transformation from hydrophilic to hydrophobic above low critical solution temperature (LCST) is called hydrophobic hydration [68].
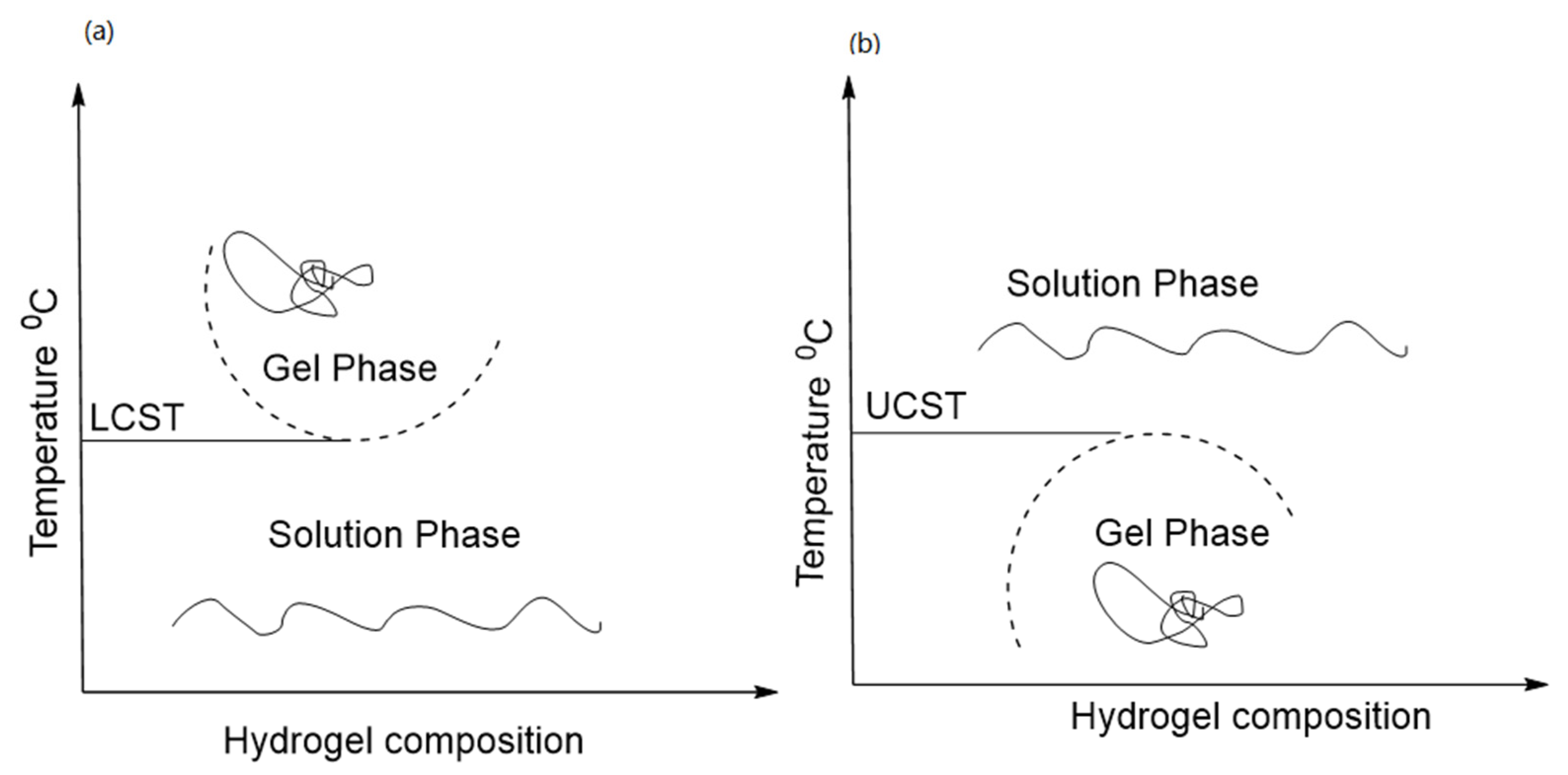
Figure 1. (a) the lower critical solution temperature (LCST) formed hydrogel on increasing temperature, and (b) the upper critical solution temperature (UCST) formed hydrogel on cooling.
3.1. LCST Polymers
In the category of LCST polymers, poly-N-isopropyl acrylamide (PNIPAAm) is a widely studied polymer. The literature reports pioneering work on physical-chemistry aspects of solution properties of PNIPAAm and LCST polymers. These are vinyl polymers synthesized by radical polymerization with secondary amide pendant groups. The lone pairs present on the oxygen atom and the lone pair of the nitrogen atom of the amide bond serve as hydrogen bond acceptors. In contrast, the hydrogen covalently attached to the nitrogen atom is a donor of hydrogen bonds. These results inter and intramolecular hydrogen bonding with water (Figure 2). LCST is a reversible phenomenon when the temperature of the polymeric system decreases, the hydrophilic-hydrophobic balance is regained for higher hydrophilicity, and the polymers become soluble [69][70]. Moreover, due to structural changes of the polymer network, polymeric systems with LCST show a coil-globule (C-G) transition. That implies that the free Gibbs energy (∆G = ∆H − T∆S) of dissolving polymer in water is negative at reduced temperatures and positive at elevated temperature from a thermodynamics standpoint. If the enthalpy of hydrogen bonding among water molecules and polymer chains (∆H) and the entropy contribution (∆S) are both negative, such behaviour is feasible. That means, if water loses entropy as it hydrates the polymer chains, the entropy term (−T∆S) will began to dominate the operation as the temperature of the solution rises, leads to a positive Gibbs free energy of mixing [71]. Phase separation will occur as a result of this. This shift is usually reversible, allowing for a rapid, reversible, thermally induced phase change. Poly (N-isopropyacrylamide) was the first to be discovered and has been the subject of the most research [68][70].
PNIPAAm copolymers showed cloud points around 32 °C, with polymer concentrations ranging from 5–30 wt.%. Polymer with LCST 32 °C easy to design formulations with properties from room temperature to 37 °C. In the scientific community, there is growing interest in incorporating polymeric blocks in PNIPAAm’s to turn PNIPAAm’s LCST towards higher temperature for exploring hyperthermia applications for targeted disease areas and drug release observed by the disruption of polymeric architecture. A polymer chemist explored this area by incorporating hydrophilic monomer moiety to shift the hydrophilic/lipophilic balance towards more hydrophilic. This results in a greater interaction with aqueous solution as well as a higher temperature required to achieve LCST for disorganization of polymer and release of internal encapsulated cargo [72]. In the literature, the PNIPAAm block copolymer-based micelle was synthesized with different hydrophobic blocks of a polymer, such as a polystyrene and poly-L-lactide (PLA), to encapsulate doxorubicin. These LCST polymeric micelles destabilize at 37–42.5 °C and release doxorubicin, unlike at 37 °C for PNIPAAm [73].
3.2. UCST Polymers
Once the solute-solute and solvent-solvent interactions overwhelm the solute-solvent interaction to yield a positive enthalpy of mixing, the UCST phase change occurs. Assume the enthalpic term (∆H) in the Gibbs free energy expression to indicate the supramolecular association of the polymer. However, the complex molecular interaction strength diminishes with rising temperature, causing the hydration component to prevail and contribute to polymer breakdown. To achieve the UCST shift greater than the theoretical LCST shift, which can lead to total insolubility, the polymer has to be hydrophilic in nature. The reaction between poly (acrylic acid) with poly(acrylamide) in water, driven by hydrogen bonding between the carboxy and amide groups, is an example of a UCST-type polymer [71][74].
These polymers are soluble above UCST and insoluble below the critical temperature. Unlike LCST polymers, UCST polymers are less explored for aqueous systems due to some limitations, but nowadays, there is growing interest amongst the scientific community for UCST polymers. Thermoresponsive in the UCST-based copolymers depends on the hydrogen bonds or electrostatic interactions between functional groups with the surrounding aqueous environment. In this class, polymers that exhibited zwitterion and ion exhibited this phenomenon [8]. Strong supramolecular interactions (hydrogen bond, electrostatic bond in zwitterion ions) between groups of copolymers with their surrounding aqueous conditions resulted in UCST, as shown in Figure 2. poly(N-acryloyl glycinamide) polymers are non-ionic polymers shown UCST behavior in water by virtue of hydrogen bonding between the polymer side groups [75][76]. Figure 3 shows thermoresponsive behaviors in the UCST copolymers achieved by hydrogen bonds between poly(N-acryloyl glycinamide) polymer chains and electrostatic bonds between zwitterionic groups in poly(N, N′-dimethyl(methacryloylethyl)ammonium propane-sulfonate).
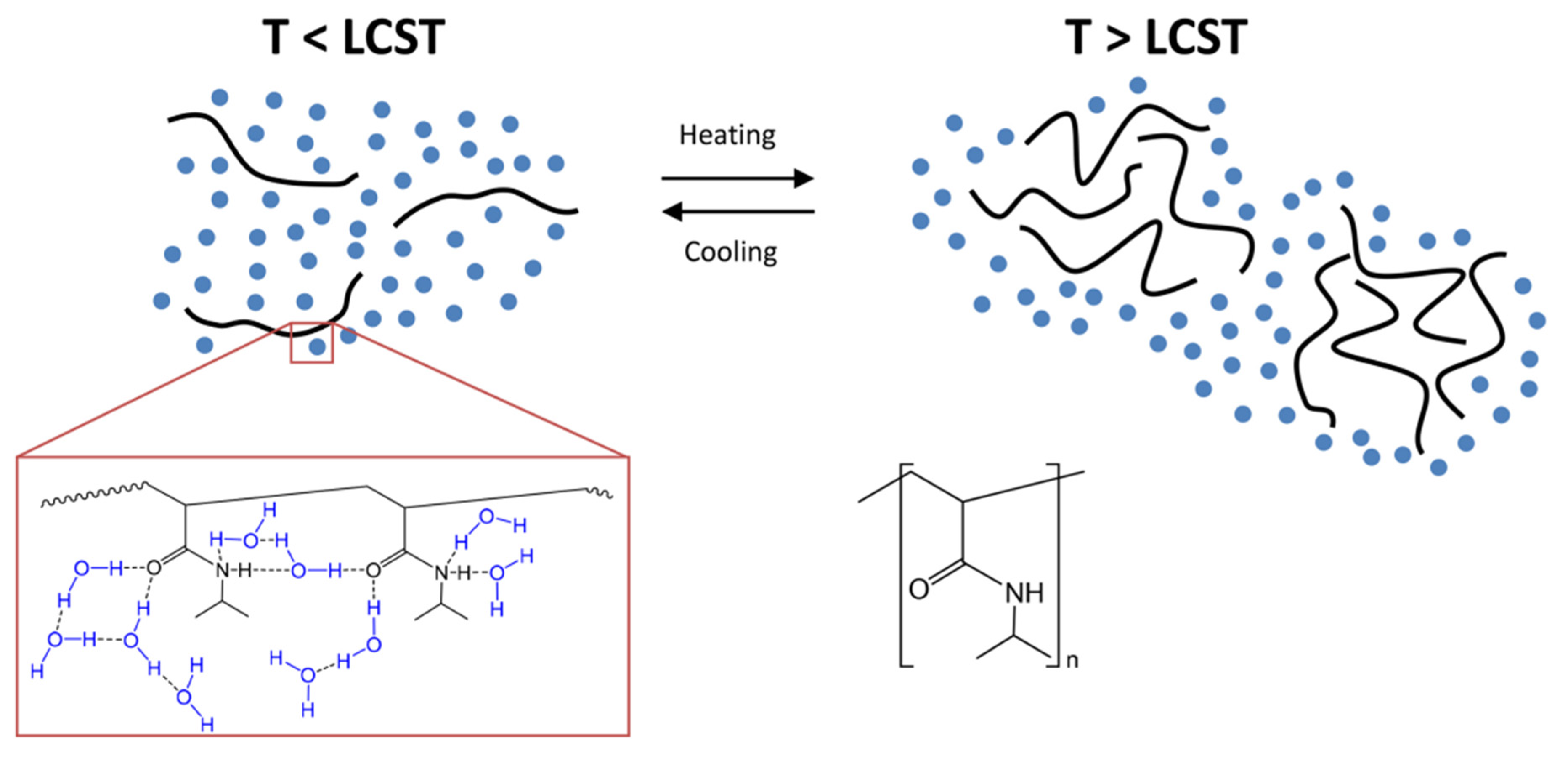
Figure 2. PNIPAAm chains (black) surrounded by water molecules (blue) as a function of temperature. Bottom-right: chemical structure of PNIPAAm. The red inset shows the possible hydrogen bonds between water molecules and polymer chains. Below the LCST, polymer chains are fully hydrated and solubilized, whereas above the LCST, they interact strongly with one another, the intrachain hydrophobic effect changes the conformation of the polymer chains to a coil state, they aggregate, and phase separate from the water phase to yield a turbid suspension. Reproduced from Bordat et al. [77] with kind permission of the copyright holder, 2019, Elsevier.
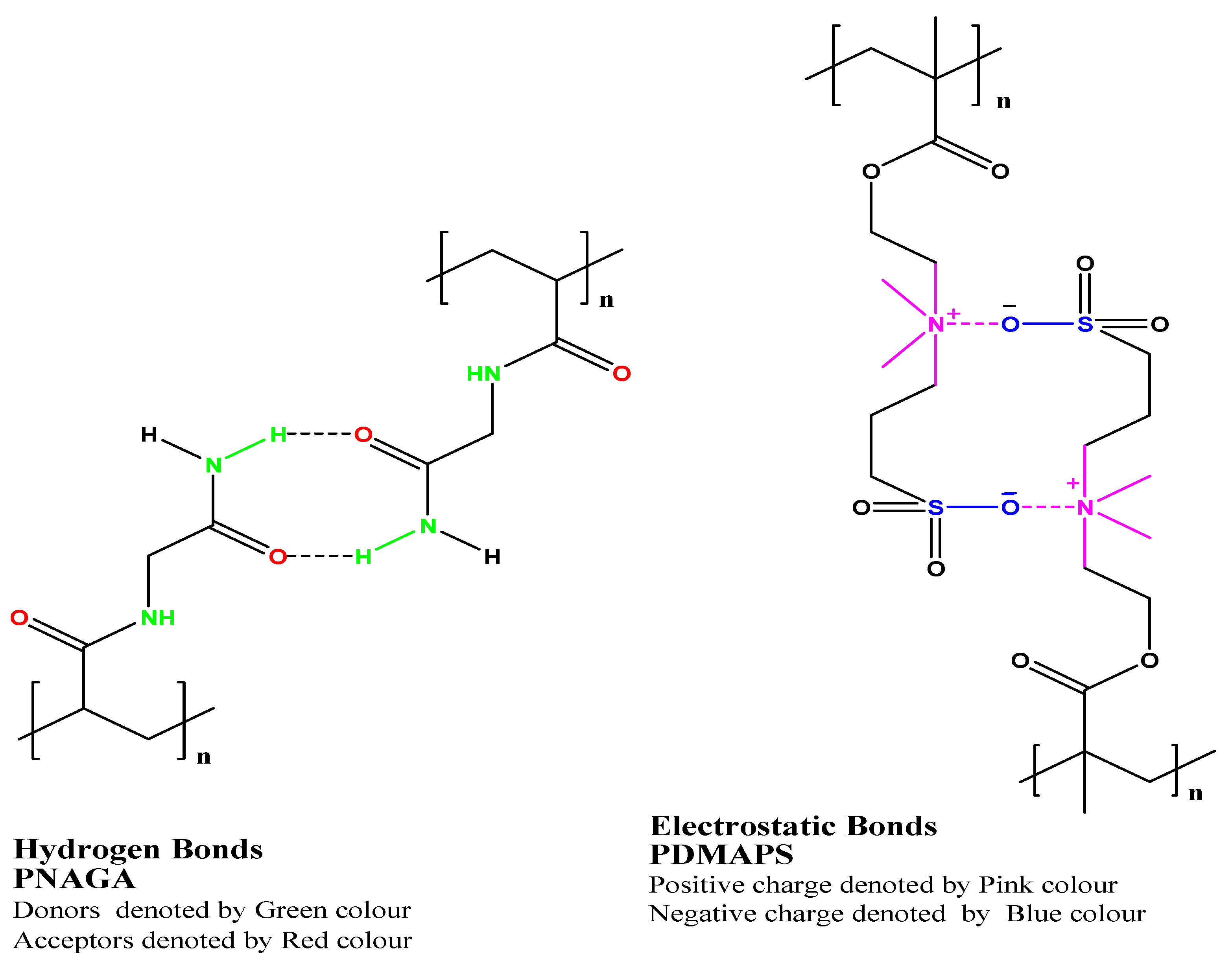
Figure 3. Thermoresponsive behaviors in the UCST copolymers are achieved by hydrogen bonds between poly(N-acryloyl glycinamide) polymer chains and electrostatic bonds between zwitterionic groups in poly(N, N′-dimethyl(methacryloylethyl)ammonium propane-sulfonate). Reproduced from Niskanen et al. [42], with the kind permission of the copyright holder, Royal Society of Chemistry, an open access article distributed under the Creative Commons Attribution License that permits unrestricted use, distribution, and reproduction in any medium.
3.3. Mechanism of Phase Transition in Thermoresponsive Hydrogels
Multiple mechanisms reported in the literature for thermoresponsive UCST and LCST copolymers resulted from phase transition in an aqueous environment. The primary mechanism is the hydration of UCST/LCST copolymers by intra and intermolecular hydrogen bonding, which affects the solubility of copolymers resulting in a change in information of hydrogels with respect to change in temperature. Factors responsible for phase transition in the thermoresponsive hydrogels are ionic interactions (electrolytes), Van-der-Waals interactions, hydrophobic interactions and hydrogen bonding. Other factors contributing to phase transition are the interaction of the functional group of copolymer with the surrounding environment, thermodynamics, and the negative energy of the system [78][79].
References
- Huang, H.; Qi, X.; Chen, Y.; Wu, Z. Thermo-Sensitive Hydrogels for Delivering Biotherapeutic Molecules: A Review. Saudi Pharm. J. 2019, 27, 990–999.
- Naik, J.B.; Pardeshi, S.R.; Patil, R.P.; Patil, P.B.; Mujumdar, A. Mucoadhesive Micro-/Nano Carriers in Ophthalmic Drug Delivery: An Overview. BioNanoScience 2020, 10, 564–582.
- Sikdar, P.; Uddin, M.M.; Dip, T.M.; Islam, S.; Hoque, M.S.; Dhar, A.K.; Wu, S. Recent Advances in the Synthesis of Smart Hydrogels. Mater. Adv. 2021, 2, 4532–4573.
- Komatsu, S.; Asoh, T.-A.; Ishihara, R.; Kikuchi, A. Fabrication of Thermoresponsive Degradable Hydrogel Made by Radical Polymerization of 2-Methylene-1,3-Dioxepane: Unique Thermal Coacervation in Hydrogel. Polymer 2019, 179, 121633.
- Matanović, M.R.; Kristl, J.; Grabnar, P.A. Thermoresponsive Polymers: Insights into Decisive Hydrogel Characteristics, Mechanisms of Gelation, and Promising Biomedical Applications. Int. J. Pharm. 2014, 472, 262–275.
- Klouda, L.; Mikos, A.G. Thermoresponsive Hydrogels in Biomedical Applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45.
- Ghaeini-Hesaroeiye, S.; Razmi Bagtash, H.; Boddohi, S.; Vasheghani-Farahani, E.; Jabbari, E. Thermoresponsive Nanogels Based on Different Polymeric Moieties for Biomedical Applications. Gels 2020, 6, 20.
- Yin, Y.; Hu, B.; Yuan, X.; Cai, L.; Gao, H.; Yang, Q. Nanogel: A Versatile Nano-Delivery System for Biomedical Applications. Pharmaceutics 2020, 12, 290.
- Kozuch, O.; Mayer, V. Pig Kidney Epithelial (PS) Cells: A Perfect Tool for the Study of Flaviviruses and Some Other Arboviruses. Acta Virol 1975, 19, 498.
- Ranganathan, N.; Joseph Bensingh, R.; Abdul Kader, M.; Nayak, S.K. Synthesis and Properties of Hydrogels Prepared by Various Polymerization Reaction Systems. In Cellulose-Based Superabsorbent Hydrogels. Polymers and Polymeric Composites: A Reference Series; Mondal, M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–25.
- Tang, S.; Floy, M.; Bhandari, R.; Sunkara, M.; Morris, A.J.; Dziubla, T.D.; Hilt, J.Z. Synthesis and Characterization of Thermoresponsive Hydrogels Based on N -Isopropylacrylamide Crosslinked with 4,4′-Dihydroxybiphenyl Diacrylate. ACS Omega 2017, 2, 8723–8729.
- Cavalu, S.; Banica, F.; Gruian, C.; Vanea, E.; Goller, G.; Simon, V. Microscopic and spectroscopic investigation of bioactive glasses for antibiotic controlled release. J. Mol. Struct. 2013, 1040, 47–52.
- Doberenz, F.; Zeng, K.; Willems, C.; Zhang, K.; Groth, T. Thermoresponsive Polymers and Their Biomedical Application in Tissue Engineering-A Review. J. Mater. Chem. B 2020, 8, 607–628.
- Chatterjee, S.; Hui, P.C. Review of Applications and Future Prospects of Stimuli-Responsive Hydrogel Based on Thermo-Responsive Biopolymers in Drug Delivery Systems. Polymers 2021, 13, 2086.
- Zhang, W.; Wang, R.; Sun, Z.; Zhu, X.; Zhao, Q.; Zhang, T.; Cholewinski, A.; Yang, F. (Kuo); Zhao, B.; Pinnaratip, R.; et al. Catechol-Functionalized Hydrogels: Biomimetic Design, Adhesion Mechanism, and Biomedical Applications. Chem. Soc. Rev. 2020, 49, 433–464.
- Pramanik, S.; Mohanto, S.; Manne, R.; Rajendran, R.R.; Deepak, A.; Edapully, S.J.; Patil, T.; Katari, O. Nanoparticle-Based Drug Delivery System: The Magic Bullet for the Treatment of Chronic Pulmonary Diseases. Mol. Pharm. 2021, 18, 3671–3718.
- Wang, Y.; Huang, F.; Chen, X.; Wang, X.-W.; Zhang, W.-B.; Peng, J.; Li, J.; Zhai, M. Stretchable, Conductive, and Self-Healing Hydrogel with Super Metal Adhesion. Chem. Mater. 2018, 30, 4289–4297.
- Hua, M.; Wu, D.; Wu, S.; Ma, Y.; Alsaid, Y.; He, X. 4D Printable Tough and Thermoresponsive Hydrogels. ACS Appl. Mater. Interfaces 2021, 13, 12689–12697.
- Shou, Y. 4D-Printable Thermoresponsive Hydrogel Exhibits High Mechanical Properties. MRS Bull. 2021, 46, 301.
- Bovone, G.; Dudaryeva, O.Y.; Marco-Dufort, B.; Tibbitt, M.W. Engineering Hydrogel Adhesion for Biomedical Applications via Chemical Design of the Junction. ACS Biomater. Sci. Eng. 2021, 7, 4048–4076.
- Rahimnejad, M.; Zhong, W. Mussel-Inspired Hydrogel Tissue Adhesives for Wound Closure. RSC Adv. 2017, 7, 47380–47396.
- Waite, J.H.; Tanzer, M.L. Polyphenolic Substance of Mytilus Edulis: Novel Adhesive Containing L-Dopa and Hydroxyproline. Science 1981, 212, 1038–1040.
- Kurakula, M.; Naveen, N.R.; Patel, B.; Manne, R.; Patel, D.B. Preparation, Optimization and Evaluation of Chitosan-Based Avanafil Nanocomplex Utilizing Antioxidants for Enhanced Neuroprotective Effect on PC12 Cells. Gels 2021, 7, 96.
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging Applications of Stimuli-Responsive Polymer Materials. Nat. Mater. 2010, 9, 101–113.
- Shi, Y.; Peng, L.; Yu, G. Nanostructured Conducting Polymer Hydrogels for Energy Storage Applications. Nanoscale 2015, 7, 12796–12806.
- Ge, D.; Lee, E.; Yang, L.; Cho, Y.; Li, M.; Gianola, D.S.; Yang, S. A Robust Smart Window: Reversibly Switching from High Transparency to Angle-Independent Structural Color Display. Adv. Mater. 2015, 27, 2489–2495.
- Strozyk, M.S.; Chanana, M.; Pastoriza-Santos, I.; Pérez-Juste, J.; Liz-Marzán, L.M. Stimuli-Responsive Materials: Protein/Polymer-Based Dual-Responsive Gold Nanoparticles with PH-Dependent Thermal Sensitivity (Adv. Funct. Mater. 7/2012). Adv. Funct. Mater. 2012, 22, 1322.
- Owusu-Nkwantabisah, S.; Gillmor, J.; Switalski, S.; Mis, M.R.; Bennett, G.; Moody, R.; Antalek, B.; Gutierrez, R.; Slater, G. Synergistic Thermoresponsive Optical Properties of a Composite Self-Healing Hydrogel. Macromolecules 2017, 50, 3671–3679.
- Han, F.; Soeriyadi, A.H.; Vivekchand, S.R.C.; Gooding, J.J. Simple Method for Tuning the Optical Properties of Thermoresponsive Plasmonic Nanogels. ACS Macro Lett. 2016, 5, 626–630.
- Cui, Z.; Lee, B.H.; Pauken, C.; Vernon, B.L. Degradation, Cytotoxicity, and Biocompatibility of NIPAAm-Based Thermosensitive, Injectable, and Bioresorbable Polymer Hydrogels. J. Biomed. Mater. Res. 2011, 98A, 159–166.
- Turabee, M.H.; Jeong, T.H.; Ramalingam, P.; Kang, J.H.; Ko, Y.T. N,N,N-Trimethyl Chitosan Embedded in Situ Pluronic F127 Hydrogel for the Treatment of Brain Tumor. Carbohydr. Polym. 2019, 203, 302–309.
- Zhang, K.; Xue, K.; Loh, X.J. Thermo-Responsive Hydrogels: From Recent Progress to Biomedical Applications. Gels 2021, 7, 77.
- Sannino, A.; Demitri, C.; Madaghiele, M. Biodegradable cellulose-based hydrogels: Design and applications. Materials 2009, 2, 353–373.
- Liu, W.; Zhang, B.; Lu, W.W.; Li, X.; Zhu, D.; De Yao, K.; Wang, Q.; Zhao, C.; Wang, C. A Rapid Temperature-Responsive Sol-Gel Reversible Poly(N-Isopropylacrylamide)-g-Methylcellulose Copolymer Hydrogel. Biomaterials 2004, 25, 3005–3012.
- Stabenfeldt, S.E.; García, A.J.; LaPlaca, M.C. Thermoreversible Laminin-Functionalized Hydrogel for Neural Tissue Engineering. J. Biomed. Mater. Res. A 2006, 77A, 718–725.
- Bhattarai, N.; Matsen, F.A.; Zhang, M. PEG-Grafted Chitosan as an Injectable Thermoreversible Hydrogel. Macromol. Biosci. 2005, 5, 107–111.
- Chen, K.-Y.; Zeng, S.-Y. Fabrication of Quaternized Chitosan Nanoparticles Using Tripolyphosphate/Genipin Dual Cross-Linkers as a Protein Delivery System. Polymers 2018, 10, 1226.
- Chen, J.-P.; Cheng, T.-H. Thermo-Responsive Chitosan-Graft-Poly(N-Isopropylacrylamide) Injectable Hydrogel for Cultivation of Chondrocytes and Meniscus Cells. Macromol. Biosci. 2006, 6, 1026–1039.
- Crompton, K.E.; Goud, J.D.; Bellamkonda, R.V.; Gengenbach, T.R.; Finkelstein, D.I.; Horne, M.K.; Forsythe, J.S. Polylysine-Functionalised Thermoresponsive Chitosan Hydrogel for Neural Tissue Engineering. Biomaterials 2007, 28, 441–449.
- Bölgen, N.; Aguilar, M.R.; Fernández, M. del M.; Gonzalo-Flores, S.; Villar-Rodil, S.; San Román, J.; Pişkin, E. Thermoresponsive Biodegradable HEMA-Lactate-Dextran-Co-NIPA Cryogels for Controlled Release of Simvastatin. Artif. Cells Nanomed. Biotechnol. 2015, 43, 40–49.
- Yang, P.; Zhu, F.; Zhang, Z.; Cheng, Y.; Wang, Z.; Li, Y. Stimuli-Responsive Polydopamine-Based Smart Materials. Chem. Soc. Rev. 2021, 50, 8319–8343.
- Niskanen, J.; Tenhu, H. How to manipulate the upper critical solution temperature (UCST)? Polym. Chem. 2017, 8, 220–232.
- Joly-Duhamel, C.; Hellio, D.; Djabourov, M. All Gelatin Networks: 1. Biodiversity and Physical Chemistry. Langmuir 2002, 18, 7208–7217.
- Yang, H.; Kao, W.J. Thermoresponsive Gelatin/Monomethoxy Poly(Ethylene GlycolI_Poly(d,l-Lactide) Hydrogels: Formulation, Characterization, and Antibacterial Drug Delivery. Pharm. Res. 2005, 23, 205–214.
- Ohya, S.; Matsuda, T. Poly(N-Isopropylacrylamide) (PNIPAM)-Grafted Gelatin as Thermoresponsive Three-Dimensional Artificial Extracellular Matrix: Molecular and Formulation Parameters vs. Cell Proliferation Potential. J. Biomater. Sci. Polym. Ed. 2005, 16, 809–827.
- Gil, E.S.; Frankowski, D.J.; Spontak, R.J.; Hudson, S.M. Swelling Behavior and Morphological Evolution of Mixed Gelatin/Silk Fibroin Hydrogels. Biomacromolecules 2005, 6, 3079–3087.
- Coughlan, D.C.; Quilty, F.P.; Corrigan, O.I. Effect of Drug Physicochemical Properties on Swelling/Deswelling Kinetics and Pulsatile Drug Release from Thermoresponsive Poly(N-Isopropylacrylamide) Hydrogels. J. Control. Release 2004, 98, 97–114.
- Na, K.; Park, J.H.; Kim, S.W.; Sun, B.K.; Woo, D.G.; Chung, H.-M.; Park, K.-H. Delivery of Dexamethasone, Ascorbate, and Growth Factor (TGF Beta-3) in Thermo-Reversible Hydrogel Constructs Embedded with Rabbit Chondrocytes. Biomaterials 2006, 27, 5951–5957.
- Liu, X.-M.; Wang, L.-S.; Wang, L.; Huang, J.; He, C. The Effect of Salt and PH on the Phase-Transition Behaviors of Temperature-Sensitive Copolymers Based on N-Isopropylacrylamide. Biomaterials 2004, 25, 5659–5666.
- Yin, X.; Hoffman, A.S.; Stayton, P.S. Poly(N-Isopropylacrylamide-Co-Propylacrylic Acid) Copolymers That Respond Sharply to Temperature and PH. Biomacromolecules 2006, 7, 1381–1385.
- Xu, F.-J.; Kang, E.-T.; Neoh, K.-G. ph- and Temperature-Responsive Hydrogels from Crosslinked Triblock Copolymers Prepared via Consecutive Atom Transfer Radical Polymerizations. Biomaterials 2006, 27, 2787–2797.
- Song, S.-C.; Lee, S.B.; Jin, J.-I.; Sohn, Y.S. A New Class of Biodegradable Thermosensitive Polymers. I. Synthesis and Characterization of Poly(Organophosphazenes) with Methoxy-Poly(Ethylene Glycol) and Amino Acid Esters as Side Groups. Macromolecules 1999, 32, 2188–2193.
- Nakayama, M.; Okano, T.; Miyazaki, T.; Kohori, F.; Sakai, K.; Yokoyama, M. Molecular Design of Biodegradable Polymeric Micelles for Temperature-Responsive Drug Release. J. Control. Release 2006, 115, 46–56.
- Hatakeyama, H.; Kikuchi, A.; Yamato, M.; Okano, T. Bio-Functionalized Thermoresponsive Interfaces Facilitating Cell Adhesion and Proliferation. Biomaterials 2006, 27, 5069–5078.
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic Block Copolymers as Novel Polymer Therapeutics for Drug and Gene Delivery. J. Control. Release 2002, 82, 189–212.
- Alexandridis, P.; Alan Hatton, T. Poly(Ethylene Oxide) poly(Propylene Oxide) poly(Ethylene Oxide) Block Copolymer Surfactants in Aqueous Solutions and at Interfaces: Thermodynamics, Structure, Dynamics, and Modeling. Colloids Surf. A 1995, 96, 1–46.
- Cortiella, J.; Nichols, J.E.; Kojima, K.; Bonassar, L.J.; Dargon, P.; Roy, A.K.; Vacant, M.P.; Niles, J.A.; Vacanti, C.A. Tissue-Engineered Lung: An in Vivo and in Vitro Comparison of Polyglycolic Acid and Pluronic F-127 Hydrogel/Somatic Lung Progenitor Cell Constructs to Support Tissue Growth. Tissue Eng. 2006, 12, 1213–1225.
- Weinand, C.; Pomerantseva, I.; Neville, C.M.; Gupta, R.; Weinberg, E.; Madisch, I.; Shapiro, F.; Abukawa, H.; Troulis, M.J.; Vacanti, J.P. Hydrogel-Beta-TCP Scaffolds and Stem Cells for Tissue Engineering Bone. Bone 2006, 38, 555–563.
- Cohn, D.; Sosnik, A.; Levy, A. Improved Reverse Thermo-Responsive Polymeric Systems. Biomaterials 2003, 24, 3707–3714.
- Cellesi, F.; Tirelli, N.; Hubbell, J.A. Towards a Fully-Synthetic Substitute of Alginate: Development of a New Process Using Thermal Gelation and Chemical Cross-Linking. Biomaterials 2004, 25, 5115–5124.
- Jeong, B.; Bae, Y.H.; Kim, S.W. In Situ Gelation of PEG-PLGA-PEG Triblock Copolymer Aqueous Solutions and Degradation Thereof. J. Biomed. Mater. Res. 2000, 50, 171–177.
- Chen, S.; Singh, J. Controlled Delivery of Testosterone from Smart Polymer Solution Based Systems: In Vitro Evaluation. Int. J. Pharm. 2005, 295, 183–190.
- Zhao, X.; Debeli, D.K.; Shan, G. A Novel Drug Loading and Release from a Thermoresponsive Hydrogel Formed in Situ Emulsion Polymerization. J. Appl. Polym. Sci. 2020, 137, 48669.
- Essawy, H.A.; Ghazy, M.B.M.; El-Hai, F.A.; Mohamed, M.F. Superabsorbent Hydrogels via Graft Polymerization of Acrylic Acid from Chitosan-Cellulose Hybrid and Their Potential in Controlled Release of Soil Nutrients. Int. J. Biol. Macromol. 2016, 89, 144–151.
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive Sol–Gel Reversible Hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162.
- Ashraf, S.; Park, H.-K.; Park, H.; Lee, S.-H. Snapshot of Phase Transition in Thermoresponsive Hydrogel PNIPAM: Role in Drug Delivery and Tissue Engineering. Macromol. Res. 2016, 24, 297–304.
- Peppas, N. Hydrogels in Pharmaceutical Formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46.
- Stetsyshyn, Y.; Raczkowska, J.; Harhay, K.; Gajos, K.; Melnyk, Y.; Dąbczyński, P.; Shevtsova, T.; Budkowski, A. Temperature-Responsive and Multi-Responsive Grafted Polymer Brushes with Transitions Based on Critical Solution Temperature: Synthesis, Properties, and Applications. Colloid Polym. Sci. 2021, 299, 363–383.
- Lu, Y.; Zhou, K.; Ding, Y.; Zhang, G.; Wu, C. Origin of Hysteresis Observed in Association and Dissociation of Polymer Chains in Water. Phys. Chem. Chem. Phys. 2010, 12, 3188.
- Najafi, M.; Habibi, M.; Fokkink, R.; Hennink, W.E.; Vermonden, T. LCST Polymers with UCST Behavior. Soft Matter 2021, 17, 2132–2141.
- Ougizawa, T.; Inoue, T. UCST and LCST Behavior in Polymer Blends and Its Thermodynamic Interpretation. Polym. J. 1986, 18, 521–527.
- Bischofberger, I.; Trappe, V. New Aspects in the Phase Behaviour of Poly-N-Isopropyl Acrylamide: Systematic Temperature Dependent Shrinking of PNiPAM Assemblies Well beyond the LCST. Sci. Rep. 2015, 5, 15520.
- Kohori, F.; Sakai, K.; Aoyagi, T.; Yokoyama, M.; Yamato, M.; Sakurai, Y.; Okano, T. Control of Adriamycin Cytotoxic Activity Using Thermally Responsive Polymeric Micelles Composed of Poly(N-Isopropylacrylamide-Co-N,N-Dimethylacrylamide)-b-Poly(d,l-Lactide). Colloids Surf. B Biointerfaces 1999, 16, 195–205.
- Cook, M.T.; Haddow, P.; Kirton, S.B.; McAuley, W.J. Polymers Exhibiting Lower Critical Solution Temperatures as a Route to Thermoreversible Gelators for Healthcare. Adv. Funct. Mater. 2021, 31, 2008123.
- Seuring, J.; Agarwal, S. Non-Ionic Homo- and Copolymers with H-Donor and H-Acceptor Units with an UCST in Water. Macromol. Chem. Phys. 2010, 211, 2109–2117.
- Seuring, J.; Agarwal, S. Polymers with Upper Critical Solution Temperature in Aqueous Solution: Unexpected Properties from Known Building Blocks. ACS Macro Lett. 2013, 2, 597–600.
- Bordat, A.; Boissenot, T.; Nicolas, J.; Tsapis, N. Thermoresponsive Polymer Nanocarriers for Biomedical Applications. Adv. Drug Deliv. Rev. 2019, 138, 167–192.
- Yu, R.; Zheng, S. Poly(Acrylic Acid)-Grafted Poly(N-Isopropyl Acrylamide) Networks: Preparation, Characterization and Hydrogel Behavior. J. Biomater. Sci., Polym. Ed. 2011, 22, 2305–2324.
- Qiu, Y.; Park, K. Environment-Sensitive Hydrogels for Drug Delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339.
More
Information
Subjects:
Materials Science, Biomaterials
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Revisions:
2 times
(View History)
Update Date:
08 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No