Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bekim Sadikovic | -- | 1767 | 2022-08-05 21:43:55 | | | |
| 2 | Dean Liu | -5 word(s) | 1762 | 2022-08-08 03:11:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rooney, K.; Sadikovic, B. DNA Methylation Episignatures in Neurodevelopmental Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/25911 (accessed on 28 July 2026).
Rooney K, Sadikovic B. DNA Methylation Episignatures in Neurodevelopmental Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/25911. Accessed July 28, 2026.
Rooney, Kathleen, Bekim Sadikovic. "DNA Methylation Episignatures in Neurodevelopmental Disorders" Encyclopedia, https://encyclopedia.pub/entry/25911 (accessed July 28, 2026).
Rooney, K., & Sadikovic, B. (2022, August 05). DNA Methylation Episignatures in Neurodevelopmental Disorders. In Encyclopedia. https://encyclopedia.pub/entry/25911
Rooney, Kathleen and Bekim Sadikovic. "DNA Methylation Episignatures in Neurodevelopmental Disorders." Encyclopedia. Web. 05 August, 2022.
Copy Citation
Large structural chromosomal deletions and duplications, referred to as copy number variants (CNVs), play a role in the pathogenesis of neurodevelopmental disorders (NDDs) through effects on gene dosage.
DNA methylation
episignature
epigenetics
copy number variant
1. Episignature Development in CNV-Associated Genomic Disorders Provides Insight into Pathological Mechanism
Changes in DNA methylation (DNAm) profiles, or episignatures, in patients with large CNV defects associated with genomic disorders have not been systematically studied, and it is plausible that large CNVs, much like gene specific variants, may exhibit unique diagnostic methylation signatures in patients with NDDs.
The group recently published findings describing episignature discovery in patients with PHMDS [1], highlighting the novel insights DNA methylation analysis can contribute to the pathogenesis of CNV disorders. PHMDS is a genomic disorder associated with deletions of chromosome 22, involving partial or whole-gene disruption of the SH3 and multiple ankyrin repeat domains 3 gene (SHANK3). Intragenic variants in SHANK3 alone are responsible for a broad range of the phenotypic features observed in PHMDS [2]. However, this gene does not explain the entire phenotype in many patients, particularly speech and motor deficits, as well as renal abnormalities. The phenotypic variability and potential involvement of additional genes within the region has been previously assessed by multiple groups [3][4]. Researchers demonstrated an episignature in patients with large deletions that was not observed in those with small deletions or SHANK3 gene level variants (Figure 1a–c). The minimal region of difference between these two deletion types, large versus small, included the bromodomain-containing protein 1 gene (BRD1), a gene involved in epigenetic mechanisms and a likely candidate gene for the methylation signature observed in these patients (Figure 1d). BRD1 is a component of a histone acetyltransferase complex that interacts with chromatin remodeling proteins and, before now, there was limited genotype–phenotype association reported in this gene. In addition, metabolic studies confirmed that these patients also exhibited very different metabolic profiles [1], further providing functional evidence for disease pathogenesis, as well as indicating targets for future therapies.
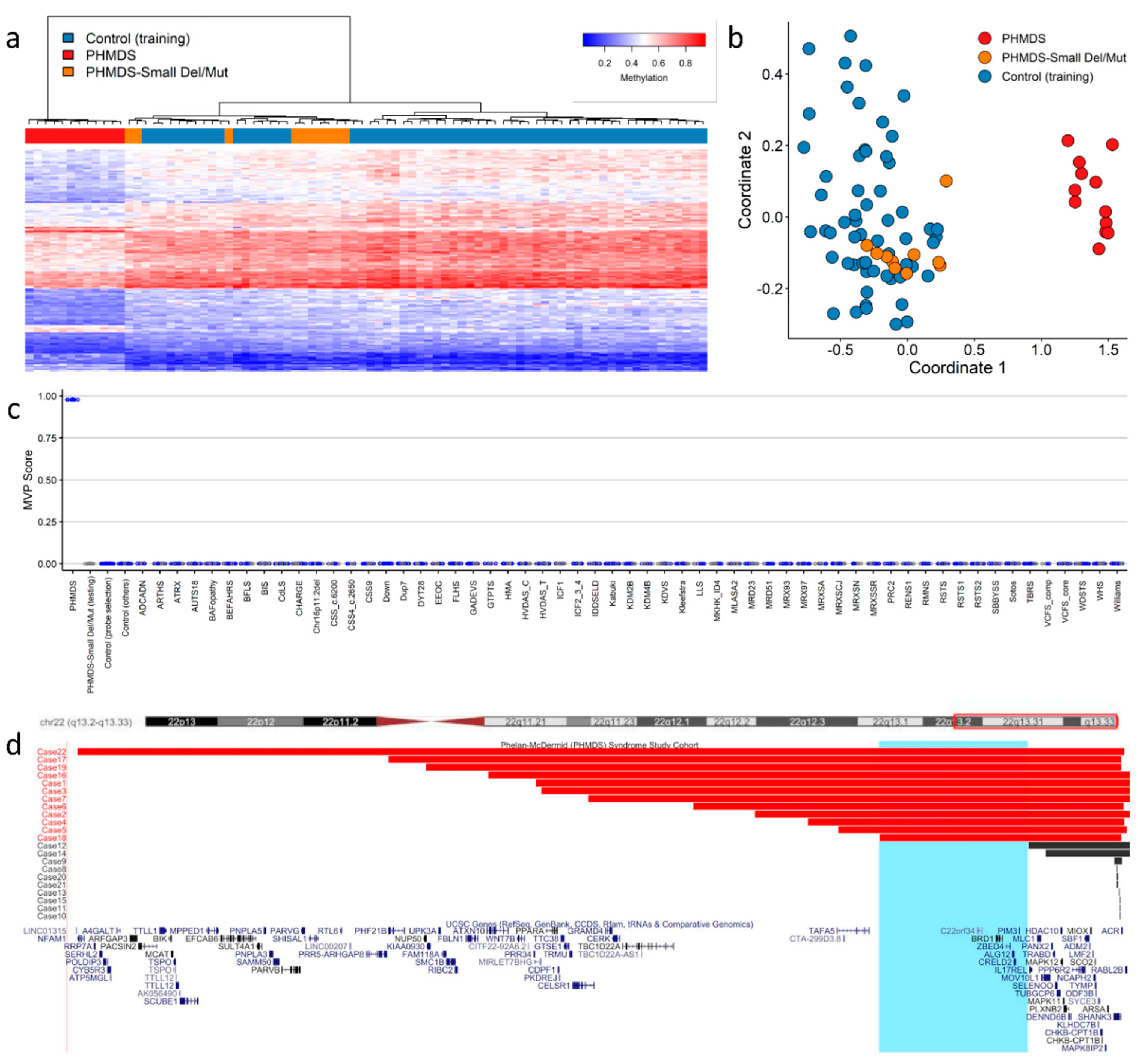
Figure 1. Phelan−McDermid syndrome (PHMDS) episignature demonstrating the critical BRD1 region: (a) Euclidean hierarchical clustering (heatmap); each column represents a single PHMDS case or control, each row represents one of the CpG probes selected for the episignature. This heatmap shows clear separation between large deletion (2−6 Mb in size) PHMDS cases (red) from controls (blue). Smaller deletions (0.01−1 Mb) and intragenic SHANK3 gene variants (Small Del/Mut) (orange) are shown to segregate with controls. (b) Multidimensional scaling (MDS) plot shows segregation of large deletion PHMDS cases from both controls and Small Del/Mut cases. (c) Support vector machine (SVM) classifier model. Model was trained using the selected probes for the PHMDS episignature, 75% of controls and 75% of other neurodevelopmental disorder samples (blue). The remaining 25% of controls and 25% of other disorder samples were used for testing (grey). Plot shows the large deletion PHMDS cases with a methylation variant pathogenicity (MVP) score close to 1 compared with all other samples, showing the specificity of the classifier and episignature. (d) PHMDS deletions illustrating the critical region of interest associated with DNA methylation episignature. The horizontal red bars represent large deletion PHMDS cases associated with the presence of a distinct episignature. The horizontal black bars represent Small Del/Mut cases that do not have a distinct DNA methylation episignature. Highlighted in light blue is the common critical region of interest (Chr22:49,228,863−50,429,645) of deletions associated with the episignature. The common region of interest contains the candidate BRD1 gene. Cytogenetic bands and known genes are presented in this figure using the UCSC genome browser [5] 2009 (GRCh37/hg19) genome build. Figure adapted with permission from Schenkel et al. [1].
2. Defined Episignatures in Other CNV-Associated Genomic Disorders Provide Rationale to Further Expand Episignature Discovery
Symmetrical dose-dependent DNAm profiles have been reported in individuals with deletion of the 7q11.23 region (Williams syndrome; WS) or duplication of the same region (7q11.23 duplication syndrome) [6], highlighting the importance of DNAm in the pathogenesis of these disorders. This region contains a number of genes associated with epigenetic mechanisms, and a study by Aref-Eshghi et al. later showed that these methylation changes resulted in unique episignatures that could differentiate WS and 7q11.23 duplication syndrome from 40 other NDDs and congenital anomaly disorders [7]. In the same study, Aref-Eshghi et al. demonstrated another example of symmetrical DNAm pattern, this time when comparing Hunter–McAlpine syndrome (HMS) and Sotos syndrome. A distinct hypermethylation episignature is observed in HMS patients with duplications involving the 5q35 region containing the NSD1 gene, a direct contrast to the robust hypomethylation episignature seen in patients with Sotos syndrome, which is the result of loss of function variants in the same NSD1 gene [7].
A DNAm signature was reported in a cohort with the genomic disorder 16p11.2 deletion syndrome (16p11.2DS) [8]—a disorder associated with a variable phenotype that includes increased susceptibility to autism spectrum disorder (ASD). Several genes within this region play a role in histone or chromatin function; however, to date, no single candidate gene has been identified to be causative of this disorder or its resultant episignature. Moreover, 16p11.2DS shows reduced penetrance and variable expressivity, and although most deletions are de novo, many are inherited from apparently unaffected parents. These so-called “susceptibility CNVs” present challenges for clinicians in counselling families [9]. Due to the presence of a cluster of low copy repeats (LCRs) in this region that mediate CNVs through non-allelic homologous recombination (NAHR), there is a reciprocal duplication disorder (16p11.2 duplication syndrome) with similar diagnostic challenges. Studying methylation changes in patients with these susceptibility CNVs and their carrier parents could potentially unlock novel insights into the role of aberrant DNAm in reduced penetrance CNV disorders.
The group recently described an aberrant DNAm pattern in patients with deletions of 12q24.31 encompassing the known histone modifier gene SET domain-containing protein 1B (SETD1B), and demonstrated that patients who harbored point mutations within SETD1B shared the same methylation episignature [10]. Larger CNVs may exhibit the same methylation affects as gene specific variants within these regions.
The most common genomic disorder is 22q11.2 deletion syndrome and is the result of a 1.5–3 Mb deletion also mediated by NAHR at a cluster of LCRs. Clinical manifestations of this disorder include DiGeorge and Velocardiofacial syndromes, and, to date, the phenotype–genotype relationship has not been fully elucidated. Through analysis of a cohort of individuals with 22q11.2 deletions, researchers identified an episignature that can differentiate 22q11.2 deletion syndrome from other NDDs on the clinical EpiSign test, including those considered in the differential diagnosis of this syndrome [11]. Among other findings, assessment of differentially methylated regions (DMRs) showed overlap with loci for orofacial clefting, a key phenotypic feature of this disorder. Through further analysis of atypical deletions and gene level variants, it may be possible to determine the gene, or genes, that play a role in the aberrant DNAm pattern observed, as well as insight into the mechanisms contributing to this disorder.
Only a few of the most prevalent genomic disorders have a candidate gene considered responsible for the entire phenotypic spectrum. Interestingly, where these candidate genes have been identified, they are predominantly involved in epigenetic regulation including chromatin remodeling or histone modification, e.g., CREBBP in Rubinstein–Taybi syndrome [12] and NSD1 in Sotos syndrome [13]. Variants in most of these genes have already been assessed for genome-wide DNAm changes, and have been shown to exhibit unique and specific episignatures [14]. Overall, the majority of CNV disorders do not have a known or suspected candidate gene of interest. However, almost all of these regions contain one or more genes with epigenetic function, e.g., chromodomain helicase DNA-binding protein 1-like (CHD1L) gene in 1q21.1 deletions and duplications, a gene that has a role in chromatin remodeling following DNA damage [15].
Taken together, the evidence suggests that CNV-associated genomic disorders may exhibit aberrant DNAm as the result of genes affected in their underlying deletions and duplications, especially when those regions include genes with epigenetic regulatory roles. CNV-associated genomic disorders are therefore strong candidates for episignature discovery. Investigating these syndromes further, including atypical CNVs and gene level variants within the same regions for possible sub-signatures, may uncover novel insights into the pathogenesis of these disorders. These studies may also identify new candidate genes responsible for some of the phenotypic presentation—should sub-signatures be uncovered for specific deleted or duplicated regions—and potentially unlock novel targets for more personalized treatment approaches.
3. Combined Detection of CNVs and DNA Methylation Episignatures in a Single Assay
Recent studies have shown it is possible to detect CNVs by applying computational methods to data obtained from DNAm arrays, such as the Illumina 450K and EPIC Bead Chip arrays [16][17][18]. Many of these pipelines are publicly available in Bioconductor, e.g., ChAMP [18][19], CopyNumber450k [20] and EpiCopy [16] (https://bioconductor.org/packages/, accessed on 19 May 2022). The ability to integrate the detection of genetic and epigenetic findings can provide a more complete view of underlying pathogenic mechanisms.
Researchers applied a similar computational approach using the DNAcopy package (Bioconductor.org) to the PHMDS cohort, and confirmed researchers could detect breakpoint coordinates similar to those obtained from conventional clinical CMA at the time of original diagnosis [1]; these findings are in line with previous studies [16][17][18][19][20].
Combining these detection methods is not without challenges, most notably in coverage of the genome, as CpG sites are not uniformly distributed throughout the genome and therefore methylation arrays lack the “backbone coverage” observed in high-density SNP arrays. However, it is plausible that, with modifications, a combined array could be developed containing a combination of copy number and CpG targeted probes to produce a clinically targeted array enabling accurate episignature and CNV analysis on a single platform. This has the potential to impact healthcare resource utilization by reducing concurrent testing in NDD patients, and decreasing the need for reflexive testing for disorders such as those associated with imprinting. There would continue to be limitations in the ability to detect low level mosaicism, as seen with existing CNV platforms; however, studies have shown the ability to detect mosaicism from methylation arrays in Kabuki syndrome 1 [21], imprinting disorders [22] and FRX [23].
Additional benefits of a combined testing platform include those to the patient; a combined array would permit screening for more disorders in a single assay, thereby potentially increasing diagnostic yield over that of the current first-tier clinical test (chromosome microarray), and shortening the time spent in the diagnostic odyssey. This approach could concurrently reduce the burden on clinical services and genetic counselling by providing results for CMA, FRX, imprinting and methylation in a single report, leading to a reduction in requisitions and clinic visits. A combined platform would also benefit oncology studies, where limitations in tumor sample availability can often impact research and diagnosis; this would permit the detection of CNVs and methylation status from the same volume of tissue as traditional testing.
References
- Schenkel, L.C.; Aref-Eshghi, E.; Rooney, K.; Kerkhof, J.; Levy, M.A.; McConkey, H.; Rogers, R.C.; Phelan, K.; Sarasua, S.M.; Jain, L.; et al. DNA methylation epi-signature is associated with two molecularly and phenotypically distinct clinical subtypes of Phelan-McDermid syndrome. Clin. Epigenetics 2021, 13, 2.
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31.
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Chen, C.-F.; Sharp, J.L.; Rollins, J.D.; Collins, J.S.; Rogers, R.C.; Phelan, K.; DuPont, B.R. 22q13.2q13.32 genomic regions associated with severity of speech delay, developmental delay, and physical features in Phelan–McDermid syndrome. Genet. Med. 2014, 16, 318–328.
- Wilson, H.L.; Crolla, J.A.; Walker, D.; Artifoni, L.; Dallapiccola, B.; Takano, T.; Vasudevan, P.; Huang, S.; Maloney, V.; Yobb, T.; et al. Interstitial 22q13 deletions: Genes other than SHANK3 have major effects on cognitive and language development. Eur. J. Hum. Genet. 2008, 16, 1301–1310.
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006.
- Strong, E.; Butcher, D.T.; Singhania, R.; Mervis, C.B.; Morris, C.A.; De Carvalho, D.; Weksberg, R.; Osborne, L.R. Symmetrical Dose-Dependent DNA-Methylation Profiles in Children with Deletion or Duplication of 7q11.23. Am. J. Hum. Genet. 2015, 97, 216–227.
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.-C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370.
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin. Epigenetics 2019, 11, 103.
- Kleinendorst, L.; Heuvel, L.M.V.D.; Henneman, L.; Van Haelst, M.M. Who ever heard of 16p11.2 deletion syndrome? Parents’ perspectives on a susceptibility copy number variation syndrome. Eur. J. Hum. Genet. 2020, 28, 1196–1204.
- Krzyzewska, I.M.; Maas, S.M.; Henneman, P.; Lip, K.V.D.; Venema, A.; Baranano, K.; Chassevent, A.; Aref-Eshghi, E.; Van Essen, A.J.; Fukuda, T.; et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenet. 2019, 11, 156.
- Rooney, K.; Levy, M.A.; Haghshenas, S.; Kerkhof, J.; Rogaia, D.; Tedesco, M.G.; Imperatore, V.; Mencarelli, A.; Squeo, G.M.; Di Venere, E.; et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. Int. J. Mol. Sci. 2021, 22, 8611.
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968.
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.Y.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.A.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207.
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. Hum. Genet. Genom. Adv. 2021, 3, 100075.
- Singh, H.R.; Nardozza, A.P.; Möller, I.R.; Knobloch, G.; Kistemaker, H.A.V.; Hassler, M.; Harrer, N.; Blessing, C.; Eustermann, S.; Kotthoff, C.; et al. A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Mol. Cell 2017, 68, 860–871.
- Cho, S.; Kim, H.-S.; Zeiger, M.A.; Umbricht, C.B.; Cope, L.M. Measuring DNA Copy Number Variation Using High-Density Methylation Microarrays. J. Comput. Biol. 2019, 26, 295–304.
- Feber, A.; Guilhamon, P.; Lechner, M.; Fenton, T.; Wilson, G.A.; Thirlwell, C.; Morris, T.J.; Flanagan, A.M.; Teschendorff, A.E.; Kelly, J.D.; et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 2014, 15, R30.
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984.
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430.
- Papillon-Cavanagh, S.; Fortin, J.-P.; De Jay, N. CopyNumber 450k: An R Package for CNV Inference Using Illumina 450k DNA Methylation Assay. 2013. Available online: http://rdrr.io/github/spapillon/CopyNumber450k/ (accessed on 15 May 2022).
- Montano, C.; Britton, J.F.; Harris, J.R.; Kerkhof, J.; Barnes, B.T.; Lee, J.A.; Sadikovic, B.; Sobreira, N.; Fahrner, J.A. Genome-wide DNA methylation profiling confirms a case of low-level mosaic Kabuki syndrome 1. Am. J. Med. Genet. Part A 2022, 188, 2217–2225.
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Siu, V.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. Clinical Validation of a Genome-Wide DNA Methylation Assay for Molecular Diagnosis of Imprinting Disorders. J. Mol. Diagn. 2017, 19, 848–856.
- Schenkel, L.C.; Schwartz, C.; Skinner, C.; Rodenhiser, D.I.; Ainsworth, P.J.; Pare, G.; Sadikovic, B. Clinical Validation of Fragile X Syndrome Screening by DNA Methylation Array. J. Mol. Diagn. 2016, 18, 834–841.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
735
Revisions:
2 times
(View History)
Update Date:
08 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No