 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Doris Loh | -- | 6211 | 2022-08-04 18:48:00 | | | |
| 2 | Amina Yu | + 197 word(s) | 6408 | 2022-08-08 03:46:52 | | | | |
| 3 | Amina Yu | Meta information modification | 6408 | 2022-08-08 03:48:18 | | | | |
| 4 | Amina Yu | Meta information modification | 6408 | 2022-08-08 09:58:45 | | | | |
| 5 | Amina Yu | + 136 word(s) | 6544 | 2022-09-05 04:21:45 | | | | |
| 6 | Amina Yu | + 1 word(s) | 6545 | 2022-09-05 04:24:41 | | |
Video Upload Options
Melatonin (N-acetyl-5-methoxytryptamine) is a ubiquitous, mitochondria-targeted molecule present in all tested eukarya and bacteria. In March 2022, the first discovery of the serotonin N-acetyltransferase (SNAT) gene—responsible for the penultimate formation of N-acetylserotonin (NAS) before its final conversion into melatonin — in archaea further consolidates the status of melatonin as a regulator of biomolecular condensates in all three domains of life in the cellular empire. RNA viruses including SARS-CoV-2 contain proteins with intrinsically disordered regions that undergo liquid-liquid phase separation (LLPS). Liquid-liquid phase separation (LLPS) of these proteins form membraneless condensates that act as “viral factories” to facilitate and enhance replication. Phase separation of the SARS-CoV-2 nucleocapsid (N) protein is associated with mitochondrial dysfunction and rewiring of energy production away from oxidative phosphorylation (OXPHOS) to favor aerobic glycolysis in cytoplasm. Increased adenosine triphosphate (ATP) in cytoplasm supports viral replication. Melatonin protects mitochondria from damage, maintains adequate levels to disassemble “viral factories”, and prevents suppression of host antiviral immune responses by inhibiting nucleocapsid phase separation via antioxidant-dependent and -independent means.
1. Introduction
2. Melatonin Rescues Mitochondrial Membrane Potential from SARS-CoV-2 Envelope Protein-Induced Depolarization
2.1. Membrane Depolarization Impairs Oxidative Phosphorylation and Cation Homeostasis
2.2. Viroporin Ion Channel Activities May Regulate Virus Phase Separation
3. Melatonin Attenuates Membrane Depolarization and Balances Ion Homeostasis by Antioxidant-Dependent and -Independent Mechanisms to Protect Mitochondria and Lymphocytes during Viral Infection and PASC
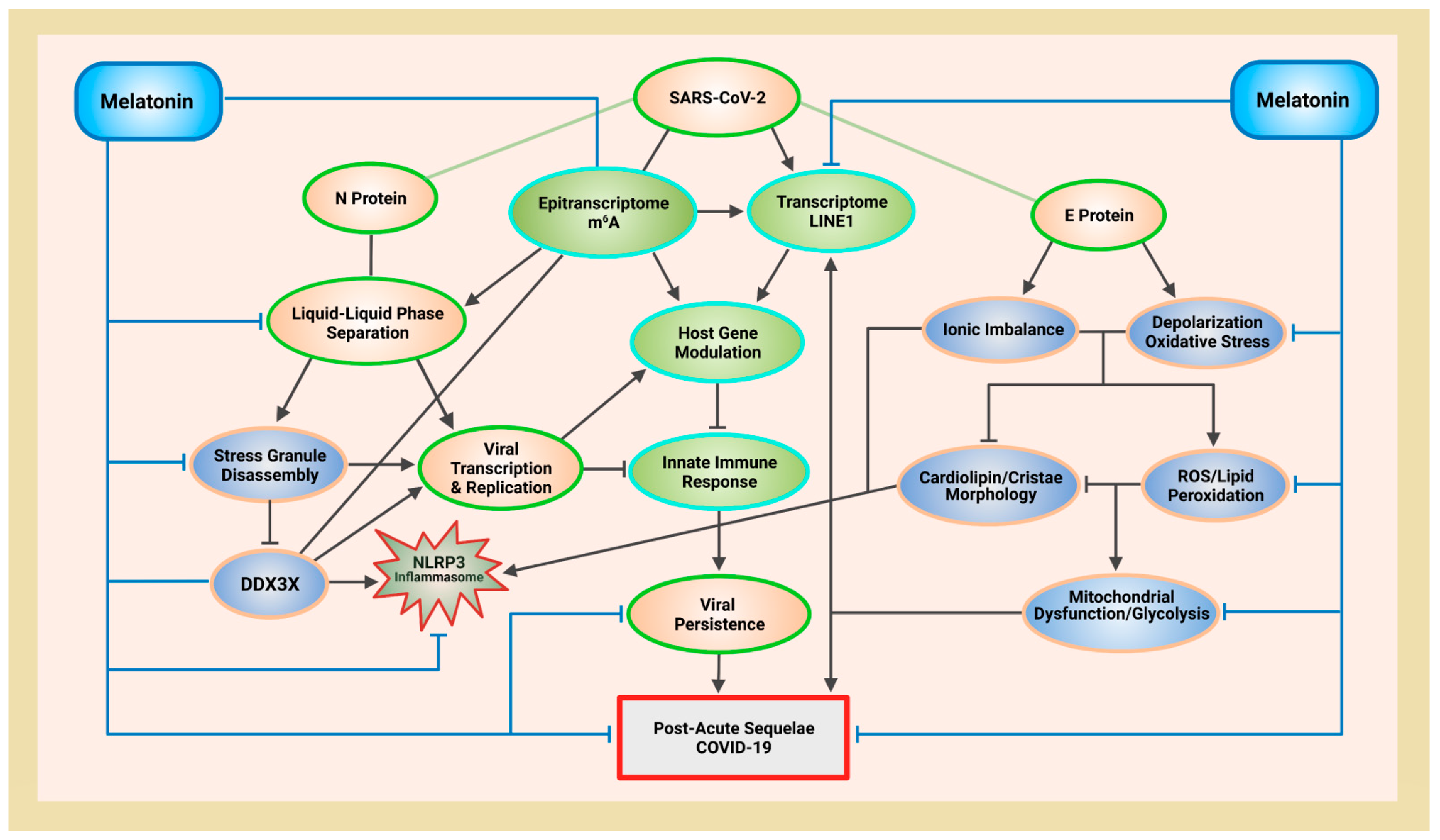
4. Melatonin Protects Mitochondria Cristae Morphology and ATP Production via Antioxidant-Dependent and -Independent Mechanisms
4.1. Melatonin Suppresses Aerobic Glycolysis to Enhance Oxidative Phosphorylation
4.2. Melatonin and Metabolites Preserve Cardiolipin Function in Cristae by Preventing Lipid Peroxidation Cascades
5. Melatonin Targets NLRP3 Inflammasomes via Cardiolipin and DDX3X
6. DDX3X Is a “Double-Edged Sword” That Mediates Host Antiviral Immunity and Viral Replication
7. N Protein Must Phase Separation to Target G3BP1 and Disassemble Stress Granules
8. The Formation of “Viral Factories” by N Protein LLPS Is Tuned by Phosphorylation
9. Melatonin Disrupts Formation of “Viral Factories” by Regulating GSK‐3 Phosphorylation of N Protein Condensates
The GSK-3 kinase is implicated in enhancing virus replication, assembly, and release [285][286][287]. As part of the innate antiviral response, GSK-3 acts as a signaling molecule that may be involved in the sensing of nucleic acids of RNA and DNA viruses. It is not only responsible for the rapid activation of type I IFN signaling cascades [288], but also serves as the crux of multiple cell signaling pathways during various stages of viral replication [289]. The activation of GSK-3 in infected cells may be responsible for increased replication and pathophysiology by promoting systemic inflammation, renal dysfunction, and hepatotoxicity via the regulation of cytokine production and cell migration [290][291], as well as the transcriptional regulation of nuclear factor kappa B (NF-κB) [292]. GSK-3 also elevates oxidative stress in infected cells by downregulating the Nrf2 and the Nrf2/antioxidant response element (ARE) pathway [293][294]. GSK-3 directly inhibits nuclear factor erythroid 2-related factor (Nrf2) activation and indirectly inhibits Nrf2 post-induction [295]. The increased oxidative stress from GSK-3 activities may induce the assembly of SGs, but more importantly, the activation of GSK-3 may actually be the elusive, underlying mechanism that is responsible for the disassembly of SGs by SARS-CoV-2 N proteins [273]. The phosphorylation of N protein by GSK-3 not only determines the viscosity and function of condensates formed by N protein LLPS, but GSK-3 can also regulate DDX3X functions to control stress granule assembly and disassembly (Figure 1). GSK-3 is responsible for the phosphorylation of Gle1A which is is recruited to SGs in the cytoplasm during stress to regulate SG dynamics, assembly, and disassembly by controlling how DDX3X binds to RNA [296][297]. Phosphorylation of Gle1A by GSK-3 alters the biochemical properties and electrophoretic mobility that allows Gle1A to bind and inhibit DDX3X ATPase activities that ultimately results in the disassembly of SGs.
Melatonin suppresses hyperphosphorylation of N protein by inhibiting the gene expression of GSK-3 and deactivating GSK-3 by promoting its phosphorylation. In Neuro2A cells subjected to okadaic acid (OA) treatment to induce phosphorylation of tau by GSK-3β exhibited elevated ROS and cytotoxicity, resulting in the loss of cell viability of up to 60%. Incubation with 200 μM melatonin for 24 h completely reversed tau-induced cytotoxicity, while at 100 μM concentration, melatonin completely restored cell viability, where melatonin effectively reduced the total mRNA expression level of GSK-3β [298].
Additionally, in human mesenchymal stem cells, melatonin attenuated adipogenic differentiation by suppressing GSK-3β activities [299]. Male Wistar rats subjected to bilateral renal ischemia to induce ischemia/reperfusion (I/R) injury showed increased lipid peroxidation and elevated lactate dehydrogenase (LDH) in plasma compared to controls. Treatment with melatonin (10 mg/kg, i.p.) 30 min before renal clamping markedly reduced lipid peroxidation and LDH levels in plasma, while the phosphorylation of GSK-3β was significantly enhanced via the restoration of AKT phosphorylation in the melatonin-treated group [300].
10. Conclusion
As COVID-19 transitions inevitably from pandemic to endemic, it is presently unclear how continued endemic infections from evolving SARS-CoV-2 variants will shape human health in the years to come. The detrimental effects of viral replication and persistence causing excess oxidative stress and mitochondrial distress can result in downstream effects that alter both host and viral RNA methylomes. Consequently, SARS-CoV-2 introduces a complex, fertile landscape that fosters a wide-array of challenging and often unexplained manifestations [301] during acute infection and post-infection. The timely application of melatonin as an essential adjuvant during acute infection and recovery can inhibit viral infection, replication, and persistence to prevent the hijacking of mitochondria and other vital host resources associated with immune evasion and suppression.
Abbreviations
| Ca2+ | calcium |
| CL | cardiolipin |
| DNA | deoxyribonucleic acid |
| EBOV | Ebola virus |
| GSK | glycogen synthase kinase |
| IBM | inner boundary membrane |
| IDR | intrinsically disordered region |
| IFN | interferon |
| IMM | inner mitochondrial membrane |
| I.P. | intraperitoneal |
| ISG | interferon-stimulated gene |
| ISR | integrated stress response |
| JAK-STAT | Janus kinase-signal transducers and activators of transcription |
| K+ | potassium ion |
| LINE1, L1 | long interspersed nuclear element 1 |
| m6A | N6-methyladenosine |
| METTL3 | methyltransferase 3 |
| METTL14 | methyltransferase 14 |
| mPTP | mitochondrial permeability transition pore |
| mRNA | messenger RNA |
| NLRP3 | NLR pyrin domain containing 3 |
| Nrf2 | nuclear factor erythroid 2-related factor |
| PBMC | peripheral blood mononuclear cells |
| PI | post-infection |
| RBP | RNA-binding protein |
| RIRR | ROS-induced ROS release |
| RNA | ribonucleic acid |
| RNA-seq | RNA sequencing |
| RNP | ribonucleoprotein |
| ROS | reactive oxygen species |
| RT | reverse transcriptase |
| RTE | retrotransposable element, retrotransposon |
| SG | stress granule |
| S/R | serine/arginine |
| TE | transposable element |
| VSV | vesicular stomatitis virus |
| YTHDF2 | YTH-domain family 2 |
| ZIKV | Zika virus |
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560.
- Brown, G.C. Control of Respiration and ATP Synthesis in Mammalian Mitochondria and Cells. Biochem. J. 1992, 284 Pt 1, 1–13.
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232.
- Khan, M.; Syed, G.H.; Kim, S.-J.; Siddiqui, A. Mitochondrial Dynamics and Viral Infections: A Close Nexus. Biochim. Biophys. Acta 2015, 1853 Pt B, 2822–2833.
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLoS Pathog. 2013, 9, e1003722.
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C Virus Triggers Mitochondrial Fission and Attenuates Apoptosis to Promote Viral Persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418.
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies-Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599.
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2021, 12, 780768.
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Residency to Host Mitochondria and Nucleolus. Cell Syst. 2020, 11, 102–108.e3.
- Pliss, A.; Kuzmin, A.N.; Prasad, P.N.; Mahajan, S.D. Mitochondrial Dysfunction: A Prelude to Neuropathogenesis of SARS-CoV-2. ACS Chem. Neurosci. 2022, 13, 308–312.
- Cortese, M.; Lee, J.-Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e5.
- Santos, A.F.; Póvoa, P.; Paixão, P.; Mendonça, A.; Taborda-Barata, L. Changes in Glycolytic Pathway in SARS-COV 2 Infection and Their Importance in Understanding the Severity of COVID-19. Front. Chem. 2021, 9, 685196.
- Ma, K.; Wu, H.; Li, P.; Li, B. LC3-II May Mediate ATR-Induced Mitophagy in Dopaminergic Neurons through SQSTM1/p62 Pathway. Acta Biochim. Biophys. Sin. 2018, 50, 1047–1061.
- Wang, S.; Zhao, Z.; Feng, X.; Cheng, Z.; Xiong, Z.; Wang, T.; Lin, J.; Zhang, M.; Hu, J.; Fan, Y.; et al. Melatonin Activates Parkin Translocation and Rescues the Impaired Mitophagy Activity of Diabetic Cardiomyopathy through Mst1 Inhibition. J. Cell. Mol. Med. 2018, 22, 5132–5144.
- da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA Viruses Promote Activation of the NLRP3 Inflammasome through Cytopathogenic Effect-Induced Potassium Efflux. Cell Death Dis. 2019, 10, 346.
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153.
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza Virus Activates Inflammasomes via Its Intracellular M2 Ion Channel. Nat. Immunol. 2010, 11, 404–410.
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287.
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus Virulence Genes with Main Focus on SARS-CoV Envelope Gene. Virus Res. 2014, 194, 124–137.
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and Inflammasomes: A Key to Understand Virus-Induced Inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738.
- McClenaghan, C.; Hanson, A.; Lee, S.-J.; Nichols, C.G. Coronavirus Proteins as Ion Channels: Current and Potential Research. Front. Immunol. 2020, 11, 573339.
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural Insights into SARS-CoV-2 Proteins. J. Mol. Biol. 2021, 433, 166725.
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130.
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and Drug Binding of the SARS-CoV-2 Envelope Protein Transmembrane Domain in Lipid Bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208.
- Breitinger, U.; Ali, N.K.M.; Sticht, H.; Breitinger, H.-G. Inhibition of SARS CoV Envelope Protein by Flavonoids and Classical Viroporin Inhibitors. Front. Microbiol. 2021, 12, 692423.
- Rizwan, T.; Kothidar, A.; Meghwani, H.; Sharma, V.; Shobhawat, R.; Saini, R.; Vaishnav, H.K.; Singh, V.; Pratap, M.; Sihag, H.; et al. Comparative Analysis of SARS-CoV-2 Envelope Viroporin Mutations from COVID-19 Deceased and Surviving Patients Revealed Implications on Its Ion-Channel Activities and Correlation with Patient Mortality. J. Biomol. Struct. Dyn. 2021, 1–16.
- Cao, Y.; Yang, R.; Wang, W.; Lee, I.; Zhang, R.; Zhang, W.; Sun, J.; Xu, B.; Meng, X. Computational Study of the Ion and Water Permeation and Transport Mechanisms of the SARS-CoV-2 Pentameric E Protein Channel. Front. Mol. Biosci 2020, 7, 565797.
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and Functional Characterization of the Membrane Association and Membrane Permeabilizing Activity of the Severe Acute Respiratory Syndrome Coronavirus Envelope Protein. Virology 2006, 349, 264–275.
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel. PLoS Pathog. 2009, 5, e1000511.
- Mehregan, A.; Pérez-Conesa, S.; Zhuang, Y.; Elbahnsi, A.; Pasini, D.; Lindahl, E.; Howard, R.J.; Ulens, C.; Delemotte, L. Probing Effects of the SARS-CoV-2 E Protein on Membrane Curvature and Intracellular Calcium. Biochim. Biophys. Acta Biomembr. 2022, 1864, 183994.
- Nardacci, R.; Colavita, F.; Castilletti, C.; Lapa, D.; Matusali, G.; Meschi, S.; Del Nonno, F.; Colombo, D.; Capobianchi, M.R.; Zumla, A.; et al. Evidences for Lipid Involvement in SARS-CoV-2 Cytopathogenesis. Cell Death Dis. 2021, 12, 263.
- Wang, P.; Luo, R.; Zhang, M.; Wang, Y.; Song, T.; Tao, T.; Li, Z.; Jin, L.; Zheng, H.; Chen, W.; et al. A Cross-Talk between Epithelium and Endothelium Mediates Human Alveolar–capillary Injury during SARS-CoV-2 Infection. Cell Death Dis. 2020, 11, 1–17.
- Mannella, C.A.; Pfeiffer, D.R.; Bradshaw, P.C.; Moraru, I.I.; Slepchenko, B.; Loew, L.M.; Hsieh, C.E.; Buttle, K.; Marko, M. Topology of the Mitochondrial Inner Membrane: Dynamics and Bioenergetic Implications. IUBMB Life 2001, 52, 93–100.
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59.
- Zick, M.; Rabl, R.; Reichert, A.S. Cristae Formation-Linking Ultrastructure and Function of Mitochondria. Biochim. Biophys. Acta 2009, 1793, 5–19.
- Gilkerson, R.W.; Selker, J.M.L.; Capaldi, R.A. The Cristal Membrane of Mitochondria Is the Principal Site of Oxidative Phosphorylation. FEBS Lett. 2003, 546, 355–358.
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148.
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual Cristae within the Same Mitochondrion Display Different Membrane Potentials and Are Functionally Independent. EMBO J. 2019, 38, e101056.
- Rieger, B.; Junge, W.; Busch, K.B. Lateral pH Gradient between OXPHOS Complex IV and F(0)F(1) ATP-Synthase in Folded Mitochondrial Membranes. Nat. Commun. 2014, 5, 3103.
- Garcia, G.C.; Bartol, T.M.; Phan, S.; Bushong, E.A.; Perkins, G.; Sejnowski, T.J.; Ellisman, M.H.; Skupin, A. Mitochondrial Morphology Provides a Mechanism for Energy Buffering at Synapses. Sci. Rep. 2019, 9, 18306.
- Scott, I.D.; Nicholls, D.G. Energy Transduction in Intact Synaptosomes. Influence of Plasma-Membrane Depolarization on the Respiration and Membrane Potential of Internal Mitochondria Determined in Situ. Biochem. J. 1980, 186, 21–33.
- Mannella, C.A. Consequences of Folding the Mitochondrial Inner Membrane. Front. Physiol. 2020, 11, 536.
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial Membrane Potential Regulates Matrix Configuration and Cytochrome c Release during Apoptosis. Cell Death Differ. 2003, 10, 709–717.
- Rasola, A.; Bernardi, P. Mitochondrial Permeability Transition in Ca(2+)-Dependent Apoptosis and Necrosis. Cell Calcium 2011, 50, 222–233.
- Liesa, M. Why Does a Mitochondrion Need Its Individual Cristae to Be Functionally Autonomous? Mol. Cell Oncol. 2020, 7, 1705119.
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446.
- Twig, G.; Shirihai, O.S. The Interplay between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951.
- Medini, H.; Zirman, A.; Mishmar, D. Immune System Cells from COVID-19 Patients Display Compromised Mitochondrial-Nuclear Expression Co-Regulation and Rewiring toward Glycolysis. iScience 2021, 24, 103471.
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter Metabolic Analysis Reveals a Close Link between Attenuated Mitochondrial Bioenergetic Function and Enhanced Glycolysis Dependency in Human Tumor Cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136.
- Codo, A.C.; Davanzo, G.G.; de Brito Monteiro, L.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5.
- Aklima, J.; Onojima, T.; Kimura, S.; Umiuchi, K.; Shibata, T.; Kuraoka, Y.; Oie, Y.; Suganuma, Y.; Ohta, Y. Effects of Matrix pH on Spontaneous Transient Depolarization and Reactive Oxygen Species Production in Mitochondria. Front. Cell Dev. Biol. 2021, 9, 692776.
- Moreno Davila, H. Molecular and Functional Diversity of Voltage-Gated Calcium Channels. Ann. N. Y. Acad. Sci. 1999, 868, 102–117.
- Pitt, G.S.; Matsui, M.; Cao, C. Voltage-Gated Calcium Channels in Nonexcitable Tissues. Annu. Rev. Physiol. 2021, 83, 183–203.
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947.
- Agirre, A.; Barco, A.; Carrasco, L.; Nieva, J.L. Viroporin-Mediated Membrane Permeabilization. Pore Formation by Nonstructural Poliovirus 2B Protein. J. Biol. Chem. 2002, 277, 40434–40441.
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of Frameshifting in Alphavirus 6K Resolves a 20-Year Enigma. Virol. J. 2008, 5, 108.
- González, M.E. Vpu Protein: The Viroporin Encoded by HIV-1. Viruses 2015, 7, 4352–4368.
- To, J.; Torres, J. Viroporins in the Influenza Virus. Cells 2019, 8.
- Liao, Y.; Lescar, J.; Tam, J.P.; Liu, D.X. Expression of SARS-Coronavirus Envelope Protein in Escherichia Coli Cells Alters Membrane Permeability. Biochem. Biophys. Res. Commun. 2004, 325, 374–380.
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993.
- Larsen, H.E.; Bardsley, E.N.; Lefkimmiatis, K.; Paterson, D.J. Dysregulation of Neuronal Ca2+ Channel Linked to Heightened Sympathetic Phenotype in Prohypertensive States. J. Neurosci. 2016, 36, 8562–8573.
- Jamal, S.M.; Landers, D.B.; Hollenberg, S.M.; Turi, Z.G.; Glotzer, T.V.; Tancredi, J.; Parrillo, J.E. Prospective Evaluation of Autonomic Dysfunction in Post-Acute Sequela of COVID-19. J. Am. Coll. Cardiol. 2022.
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic Dysfunction in “Long COVID”: Rationale, Physiology and Management Strategies. Clin. Med. 2021, 21, e63–e67.
- Papadopoulou, M.; Bakola, E.; Papapostolou, A.; Stefanou, M.-I.; Gaga, M.; Zouvelou, V.; Michopoulos, I.; Tsivgoulis, G. Autonomic Dysfunction in Long-COVID Syndrome: A Neurophysiological and Neurosonology Study. J. Neurol. 2022, 1–2.
- Raj, S.R.; Arnold, A.C.; Barboi, A.; Claydon, V.E.; Limberg, J.K.; Lucci, V.-E.M.; Numan, M.; Peltier, A.; Snapper, H.; Vernino, S.; et al. Long-COVID Postural Tachycardia Syndrome: An American Autonomic Society Statement. Clin. Auton. Res. 2021, 31, 365–368.
- Bisaccia, G.; Ricci, F.; Recce, V.; Serio, A.; Iannetti, G.; Chahal, A.A.; Ståhlberg, M.; Khanji, M.Y.; Fedorowski, A.; Gallina, S. Post-Acute Sequelae of COVID-19 and Cardiovascular Autonomic Dysfunction: What Do We Know? J. Cardiovasc. Dev. Dis 2021, 8, 156.
- Chen, X.; Cao, R.; Zhong, W. Host Calcium Channels and Pumps in Viral Infections. Cells 2019, 9, 94.
- Hyser, J.M.; Estes, M.K. Pathophysiological Consequences of Calcium-Conducting Viroporins. Annu Rev. Virol. 2015, 2, 473–496.
- Berktaş, B.M.; Gökçek, A.; Hoca, N.T.; Koyuncu, A. COVID-19 Illness and Treatment Decrease Bone Mineral Density of Surviving Hospitalized Patients. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 3046–3056.
- Mandala, V.S.; Loftis, A.R.; Shcherbakov, A.A.; Pentelute, B.L.; Hong, M. Atomic Structures of Closed and Open Influenza B M2 Proton Channel Reveal the Conduction Mechanism. Nat. Struct. Mol. Biol. 2020, 27, 160–167.
- Gargan, S.; Stevenson, N.J. Unravelling the Immunomodulatory Effects of Viral Ion Channels, towards the Treatment of Disease. Viruses 2021, 13, 2165.
- Bohmwald, K.; Gálvez, N.M.S.; Andrade, C.A.; Mora, V.P.; Muñoz, J.T.; González, P.A.; Riedel, C.A.; Kalergis, A.M. Modulation of Adaptive Immunity and Viral Infections by Ion Channels. Front. Physiol. 2021, 12, 736681.
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion Channels in Innate and Adaptive Immunity. Annu. Rev. Immunol. 2015, 33, 291–353.
- Kaivola, J.; Nyman, T.A.; Matikainen, S. Inflammasomes and SARS-CoV-2 Infection. Viruses 2021, 13, 2513.
- Campbell, G.R.; To, R.K.; Hanna, J.; Spector, S.A. SARS-CoV-2, SARS-CoV-1, and HIV-1 Derived ssRNA Sequences Activate the NLRP3 Inflammasome in Human Macrophages through a Non-Classical Pathway. iScience 2021, 24, 102295.
- Causton, H.C. SARS-CoV2 Infection and the Importance of Potassium Balance. Front. Med. 2021, 8, 744697.
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 Viroporin Encoded by ORF3a Triggers the NLRP3 Inflammatory Pathway. Virology 2022, 568, 13–22.
- Wang, J.; Zhang, K.; Goyal, P.; Grewer, C. Mechanism and Potential Sites of Potassium Interaction with Glutamate Transporters. J. Gen. Physiol. 2020, 152, e202012577.
- Kozlov, A.G.; Cheng, X.; Zhang, H.; Shinn, M.K.; Weiland, E.; Nguyen, B.; Shkel, I.A.; Zytkiewicz, E.; Finkelstein, I.J.; Record, M.T., Jr.; et al. How Glutamate Promotes Liquid-Liquid Phase Separation and DNA Binding Cooperativity of E. Coli SSB Protein. J. Mol. Biol. 2022, 434, 167562.
- Rimmele, T.S.; Rocher, A.-B.; Wellbourne-Wood, J.; Chatton, J.-Y. Control of Glutamate Transport by Extracellular Potassium: Basis for a Negative Feedback on Synaptic Transmission. Cereb. Cortex 2017, 27, 3272–3283.
- Bharadwaj, S.; Singh, M.; Kirtipal, N.; Kang, S.G. SARS-CoV-2 and Glutamine: SARS-CoV-2 Triggered Pathogenesis via Metabolic Reprograming of Glutamine in Host Cells. Front. Mol. Biosci 2020, 7, 627842.
- Wang, J.; Yang, G.; Wang, X.; Wen, Z.; Shuai, L.; Luo, J.; Wang, C.; Sun, Z.; Liu, R.; Ge, J.; et al. SARS-CoV-2 Uses Metabotropic Glutamate Receptor Subtype 2 as an Internalization Factor to Infect Cells. Cell Discov. 2021, 7, 119.
- Díaz-Resendiz, K.J.G.; Benitez-Trinidad, A.B.; Covantes-Rosales, C.E.; Toledo-Ibarra, G.A.; Ortiz-Lazareno, P.C.; Girón-Pérez, D.A.; Bueno-Durán, A.Y.; Pérez-Díaz, D.A.; Barcelos-García, R.G.; Girón-Pérez, M.I. Loss of Mitochondrial Membrane Potential (ΔΨm ) in Leucocytes as Post-COVID-19 Sequelae. J. Leukoc. Biol. 2022, 112, 23–29.
- Fitzgerald-Bocarsly, P. Human Natural Interferon-Alpha Producing Cells. Pharmacol. Ther. 1993, 60, 39–62.
- Decker, P. Neutrophils and Interferon-α-Producing Cells: Who Produces Interferon in Lupus? Arthritis Res. Ther. 2011, 13, 118.
- Wong, R.S.M.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.K.; Wong, C.K.; Chan, P.K.S.; Ng, M.H.L.; Yu, L.M.; Hui, D.S.; et al. Haematological Manifestations in Patients with Severe Acute Respiratory Syndrome: Retrospective Analysis. BMJ 2003, 326, 1358–1362.
- Zou, Z.-Y.; Ren, D.; Chen, R.-L.; Yu, B.-J.; Liu, Y.; Huang, J.-J.; Yang, Z.-J.; Zhou, Z.-P.; Feng, Y.-W.; Wu, M. Persistent Lymphopenia after Diagnosis of COVID-19 Predicts Acute Respiratory Distress Syndrome: A Retrospective Cohort Study. Eur. J. Inflam. 2021, 19, 20587392211036825.
- Ghizlane, E.A.; Manal, M.; Abderrahim, E.K.; Abdelilah, E.; Mohammed, M.; Rajae, A.; Amine, B.M.; Houssam, B.; Naima, A.; Brahim, H. Lymphopenia in Covid-19: A Single Center Retrospective Study of 589 Cases. Ann. Med. Surg. 2021, 69, 102816.
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional Exhaustion of Antiviral Lymphocytes in COVID-19 Patients. Cell. Mol. Immunol. 2020, 17, 533–535.
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Deng, Y.; Weng, Z.; Yang, L. Lymphopenia Is Associated with Severe Coronavirus Disease 2019 (COVID-19) Infections: A Systemic Review and Meta-Analysis. Int. J. Infect. Dis. 2020, 96, 131–135.
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.-Q.; Wang, Q.; Miao, H. Lymphopenia Predicts Disease Severity of COVID-19: A Descriptive and Predictive Study. Signal Transduct. Target. Ther. 2020, 5, 33.
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine 2020, 55, 102763.
- Ledderose, C.; Bao, Y.; Lidicky, M.; Zipperle, J.; Li, L.; Strasser, K.; Shapiro, N.I.; Junger, W.G. Mitochondria Are Gate-Keepers of T Cell Function by Producing the ATP That Drives Purinergic Signaling. J. Biol. Chem. 2014, 289, 25936–25945.
- Desdín-Micó, G.; Soto-Heredero, G.; Mittelbrunn, M. Mitochondrial Activity in T Cells. Mitochondrion 2018, 41, 51–57.
- Feske, S.; Giltnane, J.; Dolmetsch, R.; Staudt, L.M.; Rao, A. Gene Regulation Mediated by Calcium Signals in T Lymphocytes. Nat. Immunol. 2001, 2, 316–324.
- Campello, S.; Lacalle, R.A.; Bettella, M.; Mañes, S.; Scorrano, L.; Viola, A. Orchestration of Lymphocyte Chemotaxis by Mitochondrial Dynamics. J. Exp. Med. 2006, 203, 2879–2886.
- Zhang, K.; Li, H.; Song, Z. Membrane Depolarization Activates the Mitochondrial Protease OMA1 by Stimulating Self-Cleavage. EMBO Rep. 2014, 15, 576–585.
- Jahangir, A.; Ozcan, C.; Holmuhamedov, E.L.; Terzic, A. Increased Calcium Vulnerability of Senescent Cardiac Mitochondria: Protective Role for a Mitochondrial Potassium Channel Opener. Mech. Ageing Dev. 2001, 122, 1073–1086.
- Glitsch, M.D.; Bakowski, D.; Parekh, A.B. Store-Operated Ca2+ Entry Depends on Mitochondrial Ca2+ Uptake. EMBO J. 2002, 21, 6744–6754.
- Santos, J.H.; Hunakova, L.U.; Chen, Y.; Bortner, C.; Van Houten, B. Cell Sorting Experiments Link Persistent Mitochondrial DNA Damage with Loss of Mitochondrial Membrane Potential and Apoptotic Cell Death. J. Biol. Chem. 2003, 278, 1728–1734.
- Peña, C.; Rincon, J.; Pedreanez, A.; Viera, N.; Mosquera, J. Chemotactic Effect of Melatonin on Leukocytes. J. Pineal Res. 2007, 43, 263–269.
- Hu, L.; Zhang, S.; Wen, H.; Liu, T.; Cai, J.; Du, D.; Zhu, D.; Chen, F.; Xia, C. Melatonin Decreases M1 Polarization via Attenuating Mitochondrial Oxidative Damage Depending on UCP2 Pathway in Prorenin-Treated Microglia. PLoS ONE 2019, 14, e0212138.
- Liu, Y.-J.; Ji, D.-M.; Liu, Z.-B.; Wang, T.-J.; Xie, F.-F.; Zhang, Z.-G.; Wei, Z.-L.; Zhou, P.; Cao, Y.-X. Melatonin Maintains Mitochondrial Membrane Potential and Decreases Excessive Intracellular Ca2+ Levels in Immature Human Oocytes. Life Sci. 2019, 235, 116810.
- Lançoni, R.; Celeghini, E.C.C.; Alves, M.B.R.; Lemes, K.M.; Gonella-Diaza, A.M.; Oliveira, L.Z.; de Arruda, R.P. Melatonin Added to Cryopreservation Extenders Improves the Mitochondrial Membrane Potential of Postthawed Equine Sperm. J. Equine Vet. Sci. 2018, 69, 78–83.
- Kumari, S.; Dash, D. Melatonin Elevates Intracellular Free Calcium in Human Platelets by Inositol 1,4,5-Trisphosphate Independent Mechanism. FEBS Lett. 2011, 585, 2345–2351.
- Pieri, C.; Recchioni, R.; Moroni, F.; Marcheselli, F.; Marra, M.; Marinoni, S.; Di Primio, R. Melatonin Regulates the Respiratory Burst of Human Neutrophils and Their Depolarization. J. Pineal Res. 1998, 24, 43–49.
- Fischer, T.W.; Zmijewski, M.A.; Wortsman, J.; Slominski, A. Melatonin Maintains Mitochondrial Membrane Potential and Attenuates Activation of Initiator (casp-9) and Effector Caspases (casp-3/casp-7) and PARP in UVR-Exposed HaCaT Keratinocytes. J. Pineal Res. 2008, 44, 397–407.
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.-Y. mPTP Opening Caused by Cdk5 Loss Is due to Increased Mitochondrial Ca2+ Uptake. Oncogene 2020, 39, 2797–2806.
- Park, J.; Lee, J.; Choi, C. Mitochondrial Network Determines Intracellular ROS Dynamics and Sensitivity to Oxidative Stress through Switching Inter-Mitochondrial Messengers. PLoS ONE 2011, 6, e23211.
- Niki, E. Lipid Peroxidation: Physiological Levels and Dual Biological Effects. Free Radic. Biol. Med. 2009, 47, 469–484.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438.
- Žarković, N.; Orehovec, B.; Milković, L.; Baršić, B.; Tatzber, F.; Wonisch, W.; Tarle, M.; Kmet, M.; Mataić, A.; Jakovčević, A.; et al. Preliminary Findings on the Association of the Lipid Peroxidation Product 4-Hydroxynonenal with the Lethal Outcome of Aggressive COVID-19. Antioxidants 2021, 10, 1341.
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants 2021, 10, 1483.
- Reiter, R.J.; Tan, D.-X.; Galano, A. Melatonin Reduces Lipid Peroxidation and Membrane Viscosity. Front. Physiol. 2014, 5, 377.
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.-X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective Effects of Melatonin in Reducing Oxidative Stress and in Preserving the Fluidity of Biological Membranes: A Review. J. Pineal Res. 2014, 56, 225–237.
- Petrosillo, G.; Moro, N.; Ruggiero, F.M.; Paradies, G. Melatonin Inhibits Cardiolipin Peroxidation in Mitochondria and Prevents the Mitochondrial Permeability Transition and Cytochrome c Release. Free Radic. Biol. Med. 2009, 47, 969–974.
- Livrea, M.A.; Tesoriere, L.; D’Arpa, D.; Morreale, M. Reaction of Melatonin with Lipoperoxyl Radicals in Phospholipid Bilayers. Free Radic. Biol. Med. 1997, 23, 706–711.
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal Res. 2018, 65, e12514.
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin Directly Scavenges Hydrogen Peroxide: A Potentially New Metabolic Pathway of Melatonin Biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185.
- Fischer, T.W.; Scholz, G.; Knöll, B.; Hipler, U.C.; Elsner, P. Melatonin Reduces UV-Induced Reactive Oxygen Species in a Dose-Dependent Manner in IL-3-Stimulated Leukocytes. J. Pineal Res. 2001, 31, 39–45.
- Reiter, R.J. Melatonin: Lowering the High Price of Free Radicals. News Physiol. Sci. 2000, 15, 246–250.
- Tan, D.-X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One Molecule, Many Derivatives: A Never-Ending Interaction of Melatonin with Reactive Oxygen and Nitrogen Species? J. Pineal Res. 2007, 42, 28–42.
- Vanecek, J.; Klein, D.C. Sodium-Dependent Effects of Melatonin on Membrane Potential of Neonatal Rat Pituitary Cells. Endocrinology 1992, 131, 939–946.
- Bortner, C.D.; Gomez-Angelats, M.; Cidlowski, J.A. Plasma Membrane Depolarization without Repolarization Is an Early Molecular Event in Anti-Fas-Induced Apoptosis. J. Biol. Chem. 2001, 276, 4304–4314.
- Ching, A.C.; Hughes, M.R.; Poon, A.M.; Pang, S.F. Melatonin Receptors and Melatonin Inhibition of Duck Salt Gland Secretion. Gen. Comp. Endocrinol. 1999, 116, 229–240.
- Hughes, M.R.; Kitamura, N.; Bennett, D.C.; Gray, D.A.; Sharp, P.J.; Poon, A.M.S. Effect of Melatonin on Salt Gland and Kidney Function of Gulls, Larus Glaucescens. Gen. Comp. Endocrinol. 2007, 151, 300–307.
- Farouk, S.; Al-Huqail, A.A. Sustainable Biochar And/or Melatonin Improve Salinity Tolerance in Borage Plants by Modulating Osmotic Adjustment, Antioxidants, and Ion Homeostasis. Plants 2022, 11, 765.
- Jiang, C.; Cui, Q.; Feng, K.; Xu, D.; Li, C.; Zheng, Q. Melatonin Improves Antioxidant Capacity and Ion Homeostasis and Enhances Salt Tolerance in Maize Seedlings. Acta Physiol. Plant 2016, 38, 82.
- Li, C.; Wang, P.; Wei, Z.; Liang, D.; Liu, C.; Yin, L.; Jia, D.; Fu, M.; Ma, F. The Mitigation Effects of Exogenous Melatonin on Salinity-Induced Stress in Malus Hupehensis. J. Pineal Res. 2012, 53, 298–306.
- Chakravarty, S.; Rizvi, S.I. Circadian Modulation of Sodium-Potassium ATPase and Sodium—Proton Exchanger in Human Erythrocytes: In Vitro Effect of Melatonin. Cell. Mol. Biol. 2011, 57, 80–86.
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance. Molecules 2022, 27, 705.
- Morth, J.P.; Pedersen, B.P.; Buch-Pedersen, M.J.; Andersen, J.P.; Vilsen, B.; Palmgren, M.G.; Nissen, P. A Structural Overview of the Plasma Membrane Na+,K+-ATPase and H+-ATPase Ion Pumps. Nat. Rev. Mol. Cell Biol. 2011, 12, 60–70.
- Noel, J.; Roux, D.; Pouysségur, J. Differential Localization of Na+/H+ Exchanger Isoforms (NHE1 and NHE3) in Polarized Epithelial Cell Lines. J. Cell Sci. 1996, 109 Pt 5, 929–939.
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371.
- Grinstein, S.; Rotin, D.; Mason, M.J. Na+/H+ Exchange and Growth Factor-Induced Cytosolic pH Changes. Role in Cellular Proliferation. Biochim. Biophys. Acta 1989, 988, 73–97.
- Cha, C.Y.; Oka, C.; Earm, Y.E.; Wakabayashi, S.; Noma, A. A Model of Na+/H+ Exchanger and Its Central Role in Regulation of pH and Na+ in Cardiac Myocytes. Biophys. J. 2009, 97, 2674–2683.
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434.
- Królicka, A.L.; Kruczkowska, A.; Krajewska, M.; Kusztal, M.A. Hyponatremia in Infectious Diseases-A Literature Review. Int. J. Environ. Res. Public Health 2020, 17.
- Machado, R.R.G.; Glaser, T.; Araujo, D.B.; Petiz, L.L.; Oliveira, D.B.L.; Durigon, G.S.; Leal, A.L.; Pinho, J.R.R.; Ferreira, L.C.S.; Ulrich, H.; et al. Inhibition of Severe Acute Respiratory Syndrome Coronavirus 2 Replication by Hypertonic Saline Solution in Lung and Kidney Epithelial Cells. ACS Pharmacol. Transl. Sci. 2021, 4, 1514–1527.
- Kühlbrandt, W. Biology, Structure and Mechanism of P-Type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295.
- Dalskov, S.-M.; Immerdal, L.; Niels-Christiansen, L.-L.; Hansen, G.H.; Schousboe, A.; Danielsen, E.M. Lipid Raft Localization of GABA A Receptor and Na+, K+-ATPase in Discrete Microdomain Clusters in Rat Cerebellar Granule Cells. Neurochem. Int. 2005, 46, 489–499.
- Welker, P.; Geist, B.; Frühauf, J.-H.; Salanova, M.; Groneberg, D.A.; Krause, E.; Bachmann, S. Role of Lipid Rafts in Membrane Delivery of Renal Epithelial Na+-K+-ATPase, Thick Ascending Limb. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1328–R1337.
- Fujii, T.; Takahashi, Y.; Itomi, Y.; Fujita, K.; Morii, M.; Tabuchi, Y.; Asano, S.; Tsukada, K.; Takeguchi, N.; Sakai, H. K+-Cl- Cotransporter-3a Up-Regulates Na+,K+-ATPase in Lipid Rafts of Gastric Luminal Parietal Cells. J. Biol. Chem. 2008, 283, 6869–6877.
- Pytel, E.; Olszewska-Banaszczyk, M.; Koter-Michalak, M.; Broncel, M. Increased Oxidative Stress and Decreased Membrane Fluidity in Erythrocytes of CAD Patients. Biochem. Cell Biol. 2013, 91, 315–318.
- Padmavathi, P.; Reddy, V.D.; Maturu, P.; Varadacharyulu, N. Smoking-Induced Alterations in Platelet Membrane Fluidity and Na(+)/K(+)-ATPase Activity in Chronic Cigarette Smokers. J. Atheroscler. Thromb. 2010, 17, 619–627.
- Yelinova, V.; Glazachev, Y.; Khramtsov, V.; Kudryashova, L.; Rykova, V.; Salganik, R. Studies of Human and Rat Blood under Oxidative Stress: Changes in Plasma Thiol Level, Antioxidant Enzyme Activity, Protein Carbonyl Content, and Fluidity of Erythrocyte Membrane. Biochem. Biophys. Res. Commun. 1996, 221, 300–303.
- Sutherland, E.; Dixon, B.S.; Leffert, H.L.; Skally, H.; Zaccaro, L.; Simon, F.R. Biochemical Localization of Hepatic Surface-Membrane Na+,K+-ATPase Activity Depends on Membrane Lipid Fluidity. Proc. Natl. Acad. Sci. USA 1988, 85, 8673–8677.
- García, J.J.; Reiter, R.J.; Guerrero, J.M.; Escames, G.; Yu, B.P.; Oh, C.S.; Muñoz-Hoyos, A. Melatonin Prevents Changes in Microsomal Membrane Fluidity during Induced Lipid Peroxidation. FEBS Lett. 1997, 408, 297–300.
- García, J.J.; Piñol-Ripoll, G.; Martínez-Ballarín, E.; Fuentes-Broto, L.; Miana-Mena, F.J.; Venegas, C.; Caballero, B.; Escames, G.; Coto-Montes, A.; Acuña-Castroviejo, D. Melatonin Reduces Membrane Rigidity and Oxidative Damage in the Brain of SAMP8 Mice. Neurobiol. Aging 2011, 32, 2045–2054.
- Ochoa, J.J.; Vílchez, M.J.; Palacios, M.A.; García, J.J.; Reiter, R.J.; Muñoz-Hoyos, A. Melatonin Protects against Lipid Peroxidation and Membrane Rigidity in Erythrocytes from Patients Undergoing Cardiopulmonary Bypass Surgery. J. Pineal Res. 2003, 35, 104–108.
- Bolmatov, D.; McClintic, W.T.; Taylor, G.; Stanley, C.B.; Do, C.; Collier, C.P.; Leonenko, Z.; Lavrentovich, M.O.; Katsaras, J. Deciphering Melatonin-Stabilized Phase Separation in Phospholipid Bilayers. Langmuir 2019, 35, 12236–12245.
- Santamaria, A.; Batchu, K.C.; Matsarskaia, O.; Prévost, S.F.; Russo, D.; Natali, F.; Seydel, T.; Hoffmann, I.; Laux, V.; Haertlein, M.; et al. Strikingly Different Roles of SARS-CoV-2 Fusion Peptides Uncovered by Neutron Scattering. J. Am. Chem. Soc. 2022, 144, 2968–2979.
- Deng, Y.; Angelova, A. Coronavirus-Induced Host Cubic Membranes and Lipid-Related Antiviral Therapies: A Focus on Bioactive Plasmalogens. Front. Cell Dev. Biol. 2021, 9, 630242.
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160.
- Deng, Y.; Lee, E.L.-H.; Chong, K.; Almsherqi, Z.A. Evaluation of Radical Scavenging System in Amoeba Chaos Carolinense during Nutrient Deprivation. Interface Focus 2017, 7, 20160113.
- Mannella, C.A. Structure and Dynamics of the Mitochondrial Inner Membrane Cristae. Biochim. Biophys. Acta 2006, 1763, 542–548.
- Dang, M.; Li, Y.; Song, J. ATP Biphasically Modulates LLPS of SARS-CoV-2 Nucleocapsid Protein and Specifically Binds Its RNA-Binding Domain. Biochem. Biophys. Res. Commun. 2021, 541, 50–55.
- Zheng, X.; Sun, Z.; Yu, L.; Shi, D.; Zhu, M.; Yao, H.; Li, L. Interactome Analysis of the Nucleocapsid Protein of SARS-CoV-2 Virus. Pathogens 2021, 10, 1155.
- Yu, Q.; Guo, M.; Zeng, W.; Zeng, M.; Zhang, X.; Zhang, Y.; Zhang, W.; Jiang, X.; Yu, B. Interactions between NLRP3 Inflammasome and Glycolysis in Macrophages: New Insights into Chronic Inflammation Pathogenesis. Immun. Inflamm. Dis. 2022, 10, e581.
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial Metabolic Manipulation by SARS-CoV-2 in Peripheral Blood Mononuclear Cells of Patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65.
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying Intracellular Rates of Glycolytic and Oxidative ATP Production and Consumption Using Extracellular Flux Measurements. J. Biol. Chem. 2017, 292, 7189–7207.
- Reiter, R.J.; Sharma, R.; Rosales-Corral, S. Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain Its Inhibition of Multiple Diseases. Int. J. Mol. Sci. 2021, 22, 764.
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of Mitochondrial Pyruvate Dehydrogenase Kinase: A Proposed Mechanism by Which Melatonin Causes Cancer Cells to Overcome Cytosolic Glycolysis, Reduce Tumor Biomass and Reverse Insensitivity to Chemotherapy. Melatonin Res. 2019, 2, 105–119.
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of Pyruvate Metabolism and Human Disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604.
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin Increases the Activity of the Oxidative Phosphorylation Enzymes and the Production of ATP in Rat Brain and Liver Mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357.
- Chen, X.; Hao, B.; Li, D.; Reiter, R.J.; Bai, Y.; Abay, B.; Chen, G.; Lin, S.; Zheng, T.; Ren, Y.; et al. Melatonin Inhibits Lung Cancer Development by Reversing the Warburg Effect via Stimulating the SIRT3/PDH Axis. J. Pineal Res. 2021, e12755.
- Go, G.; Yoon, Y.M.; Yoon, S.; Lee, G.; Lim, J.H.; Han, S.-Y.; Lee, S.H. Melatonin Protects Chronic Kidney Disease Mesenchymal Stem/stromal Cells against Accumulation of Methylglyoxal via Modulation of Hexokinase-2 Expression. Biomol. Ther. 2022, 30, 28.
- Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. Int. J. Mol. Sci. 2017, 18, 2098.
- Dawson, T.L.; Gores, G.J.; Nieminen, A.L.; Herman, B.; Lemasters, J.J. Mitochondria as a Source of Reactive Oxygen Species during Reductive Stress in Rat Hepatocytes. Am. J. Physiol. 1993, 264 Pt 1, C961–C967.
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kühlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 2016, 63, 445–456.
- Esparza-Perusquía, M.; Olvera-Sánchez, S.; Pardo, J.P.; Mendoza-Hernández, G.; Martínez, F.; Flores-Herrera, O. Structural and Kinetics Characterization of the F1F0-ATP Synthase Dimer. New Repercussion of Monomer-Monomer Contact. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 975–981.
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F1Fo-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607.
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Interface Mobility between Monomers in Dimeric Bovine ATP Synthase Participates in the Ultrastructure of Inner Mitochondrial Membranes. Proc. Natl. Acad. Sci. USA 2021, 118.
- Elías-Wolff, F.; Lindén, M.; Lyubartsev, A.P.; Brandt, E.G. Curvature Sensing by Cardiolipin in Simulated Buckled Membranes. Soft Matter 2019, 15, 792–802.
- Ikon, N.; Ryan, R.O. Cardiolipin and Mitochondrial Cristae Organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163.
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-Dependent Formation of Mitochondrial Respiratory Supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48.
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880.
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614.
- Agrawal, A.; Ramachandran, R. Exploring the Links between Lipid Geometry and Mitochondrial Fission: Emerging Concepts. Mitochondrion 2019, 49, 305–313.
- Parui, P.P.; Sarakar, Y.; Majumder, R.; Das, S.; Yang, H.; Yasuhara, K.; Hirota, S. Determination of Proton Concentration at Cardiolipin-Containing Membrane Interfaces and Its Relation with the Peroxidase Activity of Cytochrome C. Chem. Sci. 2019, 10, 9140–9151.
- Haines, T.H.; Dencher, N.A. Cardiolipin: A Proton Trap for Oxidative Phosphorylation. FEBS Lett. 2002, 528, 35–39.
- Afzal, N.; Lederer, W.J.; Jafri, M.S.; Mannella, C.A. Effect of Crista Morphology on Mitochondrial ATP Output: A Computational Study. Curr. Res. Physiol. 2021, 4, 163–176.
- Vähäheikkilä, M.; Peltomaa, T.; Róg, T.; Vazdar, M.; Pöyry, S.; Vattulainen, I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane? Chem. Phys. Lipids 2018, 214, 15–23.
- Claypool, S.M. Cardiolipin, a Critical Determinant of Mitochondrial Carrier Protein Assembly and Function. Biochim. Biophys. Acta 2009, 1788, 2059–2068.
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional Role of Cardiolipin in Mitochondrial Bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417.
- Shi, Y. Emerging Roles of Cardiolipin Remodeling in Mitochondrial Dysfunction Associated with Diabetes, Obesity, and Cardiovascular Diseases. J. Biomed. Res. 2010, 24, 6–15.
- Chicco, A.J.; Sparagna, G.C. Role of Cardiolipin Alterations in Mitochondrial Dysfunction and Disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial Bioenergetics and Cardiolipin Alterations in Myocardial Ischemia-Reperfusion Injury: Implications for Pharmacological Cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352.
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac Mitochondrial Energy Metabolism in Heart Failure: Role of Cardiolipin and Sirtuins. Biochim. Biophys. Acta 2016, 1861, 1544–1554.
- Han, X.; Yang, J.; Yang, K.; Zhao, Z.; Abendschein, D.R.; Gross, R.W. Alterations in Myocardial Cardiolipin Content and Composition Occur at the Very Earliest Stages of Diabetes: A Shotgun Lipidomics Study. Biochemistry 2007, 46, 6417–6428.
- Schlame, M.; Ren, M. Barth Syndrome, a Human Disorder of Cardiolipin Metabolism. FEBS Lett. 2006, 580, 5450–5455.
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of Cardiolipin in the crd1 Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function. J. Biol. Chem. 2000, 275, 22387–22394.
- Ikonomidis, I.; Lekakis, J.; Vamvakou, G.; Loizou, S.; Revela, I.; Andreotti, F.; Kremastinos, D.T.; Nihoyannopoulos, P. IgA Anticardiolipin Antibody Is Associated with the Extent of Daily-Life Ischaemia in Patients with Chronic Coronary Artery Disease. Heart 2007, 93, 1412–1413.
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and Microbiota Dysfunction in COVID-19 Pathogenesis. Mitochondrion 2020, 54, 1–7.
- Taha, M.; Samavati, L. Antiphospholipid Antibodies in COVID-19: A Meta-Analysis and Systematic Review. RMD Open 2021, 7, e001580.
- Craig, W.Y.; Poulin, S.E.; Neveux, L.M.; Palomaki, G.E.; Dostal-Johnson, D.A.; Ledue, T.B.; Ritchie, R.F. Anti-Oxidized LDL Antibodies and Antiphospholipid Antibodies in Healthy Subjects: Relationship with Lipoprotein- and Oxidation-Related Analytes. J. Autoimmun. 1995, 8, 713–726.
- Hasan Ali, O.; Bomze, D.; Risch, L.; Brugger, S.D.; Paprotny, M.; Weber, M.; Thiel, S.; Kern, L.; Albrich, W.C.; Kohler, P.; et al. Severe Coronavirus Disease 2019 (COVID-19) Is Associated With Elevated Serum Immunoglobulin (Ig) A and Antiphospholipid IgA Antibodies. Clin. Infect. Dis. 2021, 73, e2869–e2874.
- Martín-Fernández, M.; Aller, R.; Heredia-Rodríguez, M.; Gómez-Sánchez, E.; Martínez-Paz, P.; Gonzalo-Benito, H.; Sánchez-de Prada, L.; Gorgojo, Ó.; Carnicero-Frutos, I.; Tamayo, E.; et al. Lipid Peroxidation as a Hallmark of Severity in COVID-19 Patients. Redox Biol. 2021, 48, 102181.
- Petrosillo, G.; Di Venosa, N.; Pistolese, M.; Casanova, G.; Tiravanti, E.; Colantuono, G.; Federici, A.; Paradies, G.; Ruggiero, F.M. Protective Effect of Melatonin against Mitochondrial Dysfunction Associated with Cardiac Ischemia- Reperfusion: Role of Cardiolipin. FASEB J. 2006, 20, 269–276.
- Römsing, S. Development and Validation of Bioanalytical Methods: Application to Melatonin and Selected Anti-Infective Drugs. Ph.D. Thesis, Acta Universitatis Upsaliensis, Uppsala, Sweden, 2010.
- Bongiorno, D.; Ceraulo, L.; Ferrugia, M.; Filizzola, F.; Giordano, C.; Ruggirello, A.; Liveri, V.T. H-NMR and FT-IR Study of the State of Melatonin Confined in Membrane Models: Location and Interactions of Melatonin in Water Free Lecithin and AOT Reversed Micelles. ARKIVOC 2004, 2004, 251–262.
- Ceraulo, L.; Ferrugia, M.; Tesoriere, L.; Segreto, S.; Livrea, M.A.; Turco Liveri, V. Interactions of Melatonin with Membrane Models: Portioning of Melatonin in AOT and Lecithin Reversed Micelles. J. Pineal Res. 1999, 26, 108–112.
- Galano, A.; Tan, D.X.; Reiter, R.J. Cyclic 3-Hydroxymelatonin, a Key Metabolite Enhancing the Peroxyl Radical Scavenging Activity of Melatonin. RSC Adv. 2014, 4, 5220.
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-Hydroxymelatonin: A Melatonin Metabolite Generated as a Result of Hydroxyl Radical Scavenging. Biol. Signals Recept. 1999, 8, 70–74.
- Bielski, B.H.; Arudi, R.L.; Sutherland, M.W. A Study of the Reactivity of HO2/O2- with Unsaturated Fatty Acids. J. Biol. Chem. 1983, 258, 4759–4761.
- Aikens, J.; Dix, T.A. Perhydroxyl Radical (HOO.) Initiated Lipid Peroxidation. The Role of Fatty Acid Hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098.
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879.
- Repetto, M.; Semprine, J.; Boveris, A. Lipid Peroxidation: Chemical Mechanism, Biological Implications and Analytical Determination. In Lipid Peroxidation; Catala, A., Ed.; IntechOpen: Rijeka, Croatia, 2012.
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis Virus Viroporin 2B Activates NLRP3 Inflammasome. PLoS Pathog. 2012, 8, e1002857.
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome Formation in the Lungs of Patients with Fatal COVID-19. Inflamm. Res. 2021, 70, 7–10.
- Arioz, B.I.; Tarakcioglu, E.; Olcum, M.; Genc, S. The Role of Melatonin on NLRP3 Inflammasome Activation in Diseases. Antioxidants 2021, 10, 1020.
- Seoane, P.I.; Lee, B.; Hoyle, C.; Yu, S.; Lopez-Castejon, G.; Lowe, M.; Brough, D. The NLRP3-Inflammasome as a Sensor of Organelle Dysfunction. J. Cell Biol. 2020, 219, 12.
- Samir, P.; Kanneganti, T.-D. DDX3X Sits at the Crossroads of Liquid-Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020, 39, 1091–1095.
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “Prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737.
- Xia, S.; Chen, Z.; Shen, C.; Fu, T.-M. Higher-Order Assemblies in Immune Signaling: Supramolecular Complexes and Phase Separation. Protein Cell 2021, 12, 680–694.
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 Inflammasome Activation and Cell Death. Cell. Mol. Immunol. 2021, 18, 2114–2127.
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The Inflammasome: A Caspase-1-Activation Platform That Regulates Immune Responses and Disease Pathogenesis. Nat. Immunol. 2009, 10, 241–247.
- Huang, Z.; Tyurina, Y.Y.; Jiang, J.; Tokarska-Schlattner, M.; Boissan, M.; Lacombe, M.-L.; Epand, R.; Schlattner, U.; Epand, R.M.; Kagan, V.E. Externalization of Cardiolipin as an “Eat-Me” Mitophageal Signal Is Facilitated by NDPK-D. Biophys. J. 2014, 106, 184a.
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052.
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome Activation in Infected Macrophages Drives COVID-19 Pathology. Nature 2022, 606, 585–593.
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 Inflammasome by Sars-CoV-2 Envelope Protein. Sci. Rep. 2021, 11, 24432.
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518.
- Zeng, J.; Xie, X.; Feng, X.-L.; Xu, L.; Han, J.-B.; Yu, D.; Zou, Q.-C.; Liu, Q.; Li, X.; Ma, G.; et al. Specific Inhibition of the NLRP3 Inflammasome Suppresses Immune Overactivation and Alleviates COVID-19 like Pathology in Mice. EBioMedicine 2022, 75, 103803.
- Zhao, N.; Di, B.; Xu, L.-L. The NLRP3 Inflammasome and COVID-19: Activation, Pathogenesis and Therapeutic Strategies. Cytokine Growth Factor Rev. 2021, 61, 2–15.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129.
- Mitroulis, I.; Skendros, P.; Ritis, K. Targeting IL-1beta in Disease; the Expanding Role of NLRP3 Inflammasome. Eur. J. Intern. Med. 2010, 21, 157–163.
- Tőzsér, J.; Benkő, S. Natural Compounds as Regulators of NLRP3 Inflammasome-Mediated IL-1β Production. Mediat. Inflamm. 2016, 2016, 5460302.
- Yang, D.; Elner, S.G.; Bian, Z.-M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-Inflammatory Cytokines Increase Reactive Oxygen Species through Mitochondria and NADPH Oxidase in Cultured RPE Cells. Exp. Eye Res. 2007, 85, 462–472.
- Yoo, H.G.; Shin, B.A.; Park, J.S.; Lee, K.H.; Chay, K.O.; Yang, S.Y.; Ahn, B.W.; Jung, Y.D. IL-1beta Induces MMP-9 via Reactive Oxygen Species and NF-kappaB in Murine Macrophage RAW 264.7 Cells. Biochem. Biophys. Res. Commun. 2002, 298, 251–256.
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 Infection and Overactivation of Nlrp3 Inflammasome as a Trigger of Cytokine “Storm” and Risk Factor for Damage of Hematopoietic Stem Cells. Leukemia 2020, 34, 1726–1729.
- Somasekharan, S.P.; Gleave, M. SARS-CoV-2 Nucleocapsid Protein Interacts with Immunoregulators and Stress Granules and Phase Separates to Form Liquid Droplets. FEBS Lett. 2021, 595, 2872–2896.
- Park, S.H.; Lee, S.G.; Kim, Y.; Song, K. Assignment of a Human Putative RNA Helicase Gene, DDX3, to Human X Chromosome Bands p11.3-->p11.23. Cytogenet. Cell Genet. 1998, 81, 178–179.
- Vesuna, F.; Akhrymuk, I.; Smith, A.; Winnard, P.T.; Lin, S.-C.; Scharpf, R.; Kehn-Hall, K.; Raman, V. RK-33, a Small Molecule Inhibitor of Host RNA Helicase DDX3, Suppresses Multiple Variants of SARS-CoV-2. bioRxiv 2022.
- Kumar, R.; Singh, N.; Abdin, M.Z.; Patel, A.H.; Medigeshi, G.R. Dengue Virus Capsid Interacts with DDX3X-A Potential Mechanism for Suppression of Antiviral Functions in Dengue Infection. Front. Cell. Infect. Microbiol. 2017, 7, 542.
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X Inhibitors, an Effective Way to Overcome HIV-1 Resistance Targeting Host Proteins. Eur. J. Med. Chem. 2020, 200, 112319.
- Yedavalli, V.S.R.K.; Neuveut, C.; Chi, Y.-H.; Kleiman, L.; Jeang, K.-T. Requirement of DDX3 DEAD Box RNA Helicase for HIV-1 Rev-RRE Export Function. Cell 2004, 119, 381–392.
- Pène, V.; Li, Q.; Sodroski, C.; Hsu, C.-S.; Liang, T.J. Dynamic Interaction of Stress Granules, DDX3X, and IKK-α Mediates Multiple Functions in Hepatitis C Virus Infection. J. Virol. 2015, 89, 5462–5477.
- Nelson, C.; Mrozowich, T.; Gemmill, D.L.; Park, S.M.; Patel, T.R. Human DDX3X Unwinds Japanese Encephalitis and Zika Viral 5′ Terminal Regions. Int. J. Mol. Sci. 2021, 22, 413.
- Winnard, P.T., Jr.; Vesuna, F.; Raman, V. Targeting Host DEAD-Box RNA Helicase DDX3X for Treating Viral Infections. Antivir. Res. 2021, 185, 104994.
- Saito, M.; Iestamantavicius, V.; Hess, D.; Matthias, P. Monitoring Acetylation of the RNA Helicase DDX3X, a Protein Critical for Formation of Stress Granules. In RNA Remodeling Proteins: Methods and Protocols; Boudvillain, M., Ed.; Springer: New York, NY, USA, 2021; pp. 217–234.
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nature 2019, 573, 590–594.
- Cui, B.C.; Sikirzhytski, V.; Aksenova, M.; Lucius, M.D.; Levon, G.H.; Mack, Z.T.; Pollack, C.; Odhiambo, D.; Broude, E.; Lizarraga, S.B.; et al. Pharmacological Inhibition of DEAD-Box RNA Helicase 3 Attenuates Stress Granule Assembly. Biochem. Pharmacol. 2020, 182, 114280.
- Lage, S.L.; Amaral, E.P.; Hilligan, K.L.; Laidlaw, E.; Rupert, A.; Namasivayan, S.; Rocco, J.; Galindo, F.; Kellogg, A.; Kumar, P.; et al. Persistent Oxidative Stress and Inflammasome Activation in CD14highCD16- Monocytes From COVID-19 Patients. Front. Immunol. 2021, 12, 799558.
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial Dysfunction as a Driver of NLRP3 Inflammasome Activation and Its Modulation through Mitophagy for Potential Therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013.
- Favero, G.; Franceschetti, L.; Bonomini, F.; Rodella, L.F.; Rezzani, R. Melatonin as an Anti-Inflammatory Agent Modulating Inflammasome Activation. Int. J. Endocrinol. 2017, 2017, 1835195.
- Zhang, Y.; Li, X.; Grailer, J.J.; Wang, N.; Wang, M.; Yao, J.; Zhong, R.; Gao, G.F.; Ward, P.A.; Tan, D.-X.; et al. Melatonin Alleviates Acute Lung Injury through Inhibiting the NLRP3 Inflammasome. J. Pineal Res. 2016, 60, 405–414.
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458.
- Yoneyama, M.; Jogi, M.; Onomoto, K. Regulation of Antiviral Innate Immune Signaling by Stress-Induced RNA Granules. J. Biochem. 2016, 159, 279–286.
- McCormick, C.; Khaperskyy, D.A. Translation Inhibition and Stress Granules in the Antiviral Immune Response. Nat. Rev. Immunol. 2017, 17, 647–660.
- Miller, C.L. Stress Granules and Virus Replication. Future Virol. 2011, 6, 1329–1338.
- de Castro, I.F.; Volonté, L.; Risco, C. Virus Factories: Biogenesis and Structural Design. Cell. Microbiol. 2013, 15, 24–34.
- Savastano, A.; Ibáñez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid Protein of SARS-CoV-2 Phase Separates into RNA-Rich Polymerase-Containing Condensates. Nat. Commun. 2020, 11, 6041.
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 5885.
- Squeglia, F.; Romano, M.; Ruggiero, A.; Maga, G.; Berisio, R. Host DDX Helicases as Possible SARS-CoV-2 Proviral Factors: A Structural Overview of Their Hijacking Through Multiple Viral Proteins. Front. Chem. 2020, 8, 602162.
- Ciccosanti, F.; Di Rienzo, M.; Romagnoli, A.; Colavita, F.; Refolo, G.; Castilletti, C.; Agrati, C.; Brai, A.; Manetti, F.; Botta, L.; et al. Proteomic Analysis Identifies the RNA Helicase DDX3X as a Host Target against SARS-CoV-2 Infection. Antivir. Res. 2021, 190, 105064.
- Hernández-Díaz, T.; Valiente-Echeverría, F.; Soto-Rifo, R. RNA Helicase DDX3: A Double-Edged Sword for Viral Replication and Immune Signaling. Microorganisms 2021, 9, 1206.
- Valiente-Echeverría, F.; Hermoso, M.A.; Soto-Rifo, R. RNA Helicase DDX3: At the Crossroad of Viral Replication and Antiviral Immunity. Rev. Med. Virol. 2015, 25, 286–299.
- Riva, V.; Maga, G. From the Magic Bullet to the Magic Target: Exploiting the Diverse Roles of DDX3X in Viral Infections and Tumorigenesis. Future Med. Chem. 2019, 11, 1357–1381.
- Wang, W.; Jia, M.; Zhao, C.; Yu, Z.; Song, H.; Qin, Y.; Zhao, W. RNF39 Mediates K48-Linked Ubiquitination of DDX3X and Inhibits RLR-Dependent Antiviral Immunity. Sci. Adv. 2021, 7, eabe5877.
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-Box Helicase DDX3X Is a Critical Component of the TANK-Binding Kinase 1-Dependent Innate Immune Response. EMBO J. 2008, 27, 2135–2146.
- Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. DEAD/H BOX 3 (DDX3) Helicase Binds the RIG-I Adaptor IPS-1 to up-Regulate IFN-Beta-Inducing Potential. Eur. J. Immunol. 2010, 40, 940–948.
- Oshiumi, H.; Kouwaki, T.; Seya, T. Accessory Factors of Cytoplasmic Viral RNA Sensors Required for Antiviral Innate Immune Response. Front. Immunol. 2016, 7, 200.
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H.; et al. Targeting Liquid-Liquid Phase Separation of SARS-CoV-2 Nucleocapsid Protein Promotes Innate Antiviral Immunity by Elevating MAVS Activity. Nat. Cell Biol. 2021, 23, 718–732.
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell 2011, 146, 448–461.
- John, L.; Samuel, C.E. Induction of Stress Granules by Interferon and down-Regulation by the Cellular RNA Adenosine Deaminase ADAR1. Virology 2014, 454–455, 299–310.
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679.
- Parker, F.; Maurier, F.; Delumeau, I.; Duchesne, M.; Faucher, D.; Debussche, L.; Dugue, A.; Schweighoffer, F.; Tocque, B. A Ras-GTPase-Activating Protein SH3-Domain-Binding Protein. Mol. Cell. Biol. 1996, 16, 2561–2569.
- Yang, P.; Mathieu, C.; Kolaitis, R.-M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch That Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28.
- Wu, J.; Liu, W.; Gong, P. A Structural Overview of RNA-Dependent RNA Polymerases from the Flaviviridae Family. Int. J. Mol. Sci. 2015, 16, 12943–12957.
- Deater, M.; Tamhankar, M.; Lloyd, R.E. TDRD3 Is an Antiviral Restriction Factor That Promotes IFN Signaling with G3BP1. PLoS Pathog. 2022, 18, e1010249.
- Yang, W.; Ru, Y.; Ren, J.; Bai, J.; Wei, J.; Fu, S.; Liu, X.; Li, D.; Zheng, H. G3BP1 Inhibits RNA Virus Replication by Positively Regulating RIG-I-Mediated Cellular Antiviral Response. Cell Death Dis. 2019, 10, 946.
- Biswal, M.; Lu, J.; Song, J. SARS-CoV-2 Nucleocapsid Protein Targets a Conserved Surface Groove of the NTF2-like Domain of G3BP1. J. Mol. Biol. 2022, 434, 167516.
- Nabeel-Shah, S.; Lee, H.; Ahmed, N.; Burke, G.L.; Farhangmehr, S.; Ashraf, K.; Pu, S.; Braunschweig, U.; Zhong, G.; Wei, H.; et al. SARS-CoV-2 Nucleocapsid Protein Binds Host mRNAs and Attenuates Stress Granules to Impair Host Stress Response. iScience 2022, 25, 103562.
- Wang, J.; Shi, C.; Xu, Q.; Yin, H. SARS-CoV-2 Nucleocapsid Protein Undergoes Liquid-Liquid Phase Separation into Stress Granules through Its N-Terminal Intrinsically Disordered Region. Cell Discov. 2021, 7, 5.
- Lian, X.J.; Gallouzi, I.-E. Oxidative Stress Increases the Number of Stress Granules in Senescent Cells and Triggers a Rapid Decrease in p21waf1/cip1 Translation. J. Biol. Chem. 2009, 284, 8877–8887.
- Luo, L.; Li, Z.; Zhao, T.; Ju, X.; Ma, P.; Jin, B.; Zhou, Y.; He, S.; Huang, J.; Xu, X.; et al. SARS-CoV-2 Nucleocapsid Protein Phase Separates with G3BPs to Disassemble Stress Granules and Facilitate Viral Production. Sci. Bull. 2021, 66, 1194–1204.
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24.
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat. Commun. 2021, 12, 502.
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734.
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for Its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4.
- Lier, C.; Becker, S.; Biedenkopf, N. Dynamic Phosphorylation of Ebola Virus VP30 in NP-Induced Inclusion Bodies. Virology 2017, 512, 39–47.
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.D.; Becker, S. Comparison of the Transcription and Replication Strategies of Marburg Virus and Ebola Virus by Using Artificial Replication Systems. J. Virol. 1999, 73, 2333–2342.
- Nikolakaki, E.; Giannakouros, T. SR/RS Motifs as Critical Determinants of Coronavirus Life Cycle. Front. Mol. Biosci. 2020, 7, 219.
- Sarhan, M.A.; Abdel-Hakeem, M.S.; Mason, A.L.; Tyrrell, D.L.; Houghton, M. Glycogen Synthase Kinase 3β Inhibitors Prevent Hepatitis C Virus Release/assembly through Perturbation of Lipid Metabolism. Sci. Rep. 2017, 7, 1–12.
- Cuartas-López, A.M.; Gallego-Gómez, J.C. Glycogen Synthase Kinase 3ß Participates in Late Stages of Dengue Virus-2 Infection. Mem. Inst. Oswaldo Cruz 2020, 115, e190357.
- Guendel, I.; Iordanskiy, S.; Van Duyne, R.; Kehn-Hall, K.; Saifuddin, M.; Das, R.; Jaworski, E.; Sampey, G.C.; Senina, S.; Shultz, L.; et al. Novel Neuroprotective GSK-3β Inhibitor Restricts Tat-Mediated HIV-1 Replication. J. Virol. 2014, 88, 1189–1208.
- Marineau, A.; Khan, K.A.; Servant, M.J. Roles of GSK-3 and β-Catenin in Antiviral Innate Immune Sensing of Nucleic Acids. Cells 2020, 9, 897.
- Alfhili, M.A.; Alsughayyir, J.; McCubrey, J.A.; Akula, S.M. GSK-3-Associated Signaling Is Crucial to Virus Infection of Cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118767.
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595.
- Dugo, L.; Collin, M.; Allen, D.A.; Patel, N.S.A.; Bauer, I.; Mervaala, E.M.A.; Louhelainen, M.; Foster, S.J.; Yaqoob, M.M.; Thiemermann, C. GSK-3beta Inhibitors Attenuate the Organ Injury/dysfunction Caused by Endotoxemia in the Rat. Crit. Care Med. 2005, 33, 1903–1912.
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-kappaB Activation. Nature 2000, 406, 86–90.
- Chen, X.; Liu, Y.; Zhu, J.; Lei, S.; Dong, Y.; Li, L.; Jiang, B.; Tan, L.; Wu, J.; Yu, S.; et al. GSK-3β Downregulates Nrf2 in Cultured Cortical Neurons and in a Rat Model of Cerebral Ischemia-Reperfusion. Sci. Rep. 2016, 6, 20196.
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-Mediated Keap1-Independent Regulation of Nrf2 Antioxidant Response: A Molecular Rheostat of Acute Kidney Injury to Chronic Kidney Disease Transition. Redox Biol. 2019, 26, 101275.
- Culbreth, M.; Aschner, M. GSK-3β, a Double-Edged Sword in Nrf2 Regulation: Implications for Neurological Dysfunction and Disease. F1000Research 2018, 7, 1043.
- Aryanpur, P.P.; Regan, C.A.; Collins, J.M.; Mittelmeier, T.M.; Renner, D.M.; Vergara, A.M.; Brown, N.P.; Bolger, T.A. Gle1 Regulates RNA Binding of the DEAD-Box Helicase Ded1 in Its Complex Role in Translation Initiation. Mol. Cell. Biol. 2017, 37.
- Aditi; Folkmann, A.W.; Wente, S.R. Cytoplasmic hGle1A Regulates Stress Granules by Modulation of Translation. Mol. Biol. Cell 2015, 26, 1476–1490.
- Das, R.; Balmik, A.A.; Chinnathambi, S. Melatonin Reduces GSK3β-Mediated Tau Phosphorylation, Enhances Nrf2 Nuclear Translocation and Anti-Inflammation. ASN Neuro 2020, 12, 1759091420981204.
- Rhee, Y.-H.; Ahn, J.-C. Melatonin Attenuated Adipogenesis through Reduction of the CCAAT/enhancer Binding Protein Beta by Regulating the Glycogen Synthase 3 Beta in Human Mesenchymal Stem Cells. J. Physiol. Biochem. 2016, 72, 145–155.
- Hadj Ayed Tka, K.; Mahfoudh Boussaid, A.; Zaouali, M.A.; Kammoun, R.; Bejaoui, M.; Ghoul Mazgar, S.; Rosello Catafau, J.; Ben Abdennebi, H. Melatonin Modulates Endoplasmic Reticulum Stress and Akt/GSK3-Beta Signaling Pathway in a Rat Model of Renal Warm Ischemia Reperfusion. Anal. Cell. Pathol. 2015, 2015, 635172.
- Choutka, J.; Jansari, V.; Hornig, M.; Iwasaki, A. Unexplained Post-Acute Infection Syndromes. Nat. Med. 2022, 28, 911–923.

