 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raafat El-Awady | -- | 5076 | 2022-08-04 22:40:28 | | | |
| 2 | Amina Yu | Meta information modification | 5076 | 2022-08-08 03:37:22 | | |
Video Upload Options
Histone deacetylases (HDACs) are important modulators of the epigenetic constitution of cancer cells. It has become increasingly known that HDACs have the capacity to regulate various cellular systems through the deacetylation of histone and bounteous nonhistone proteins that are rooted in complex pathways in cancer cells to evade death pathways and immune surveillance. The dependency of cancer cells on divergent pathways in response to different environmental stresses has been well established. This is through triggering various molecular mechanisms that promote genomic instability and mutations, reprogram different metabolic systems, and alter gene expression patterns to escape the growth inhibitory signals and the body’s immune system inspection. A better understanding of the underlying molecular pathways involved in the adaption of cancer cells to different stressors might open a new avenue for more efficacious strategies for cancer therapy.
1. Histone Deacetylases (HDACs)
1.1. HDACs Classification
1.2. Dysregulation of HDACs in Cancer
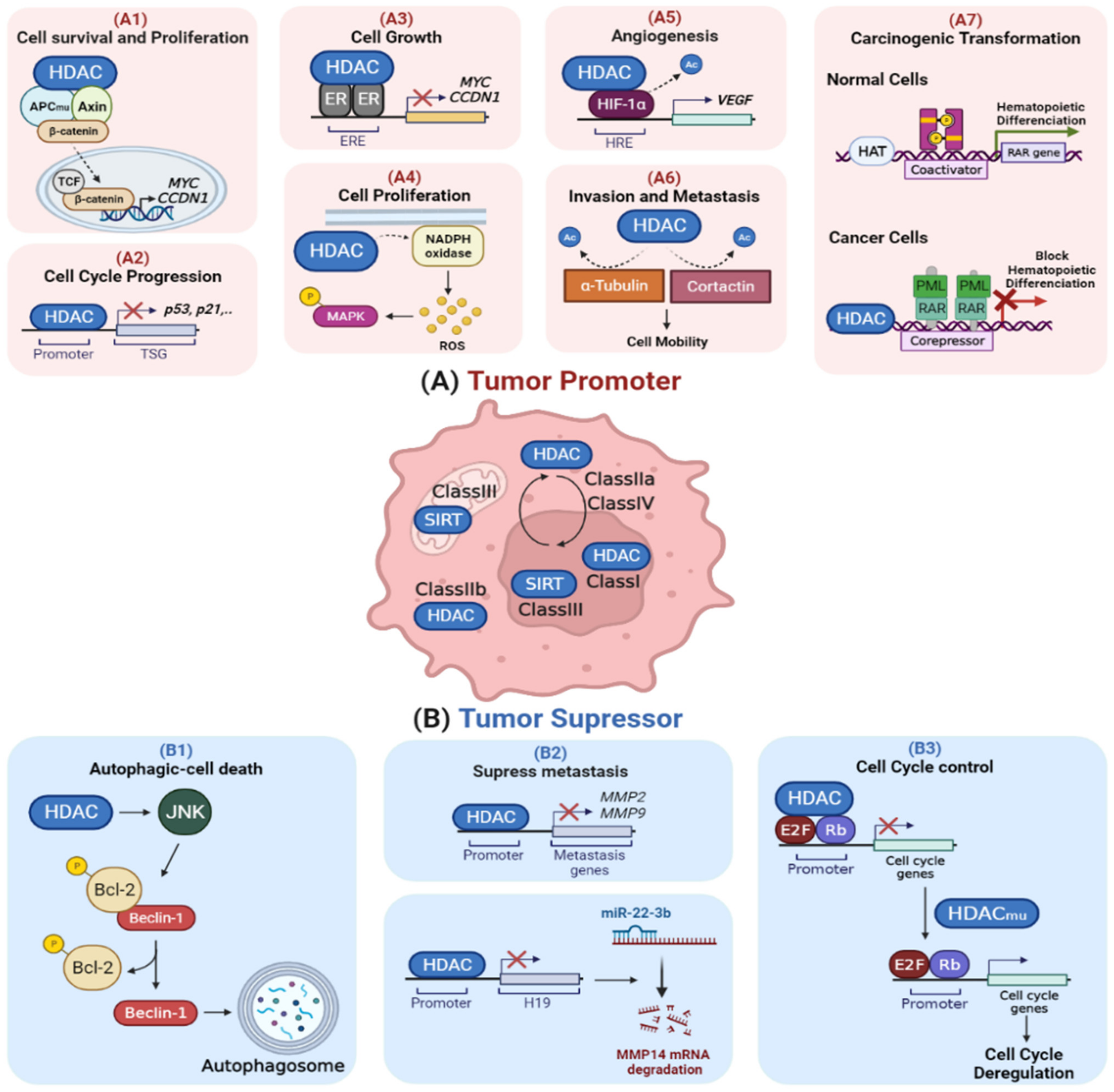
2. Role of HDACs in Cellular Stress Response
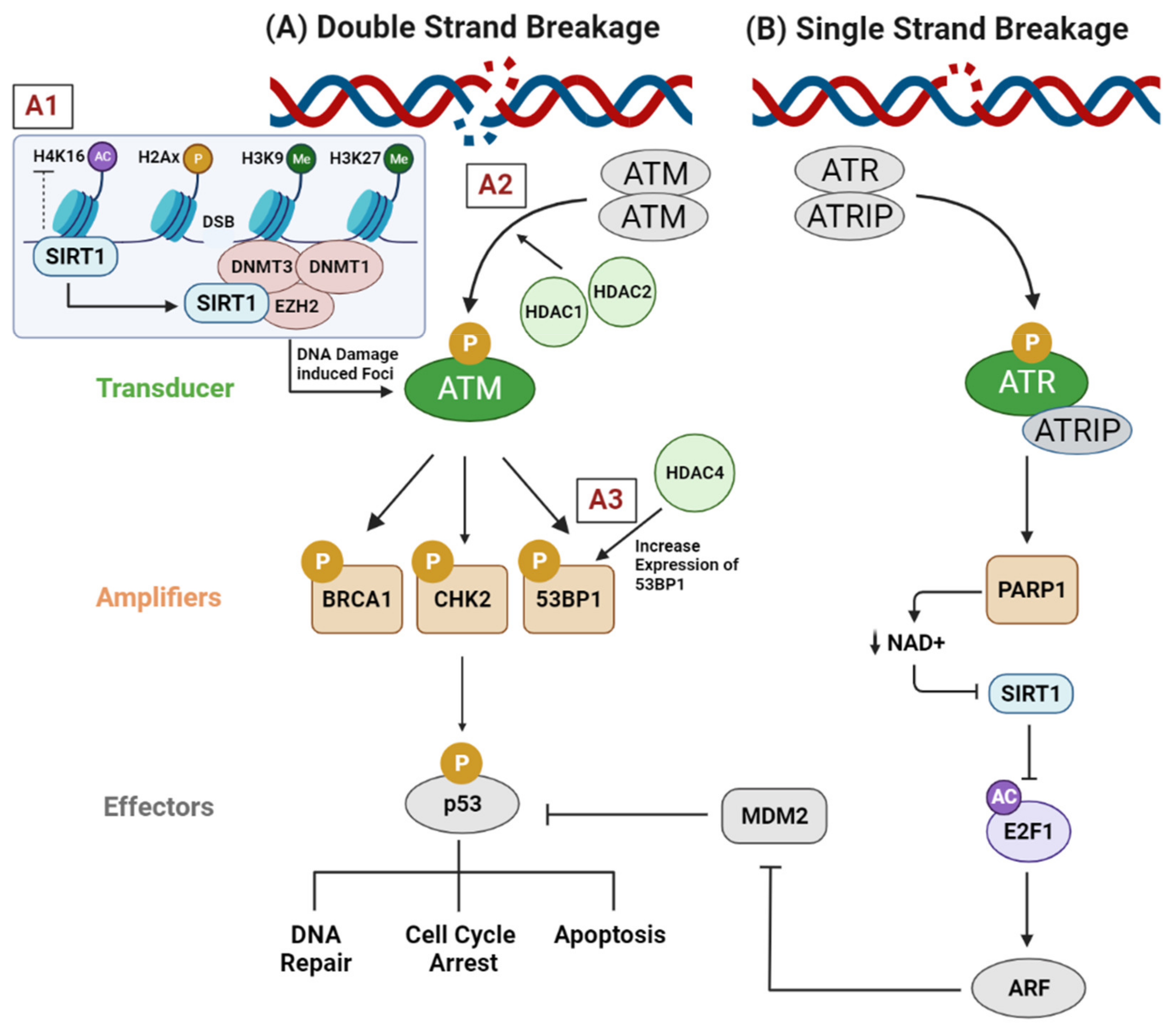
2.1. Genotoxic Stress (DNA Replication Stress, DNA Damage Response, and DNA Repair Pathways)
2.2. Proteotoxic Stress (Heat Shock Response and Endoplasmic Reticulum Stress)

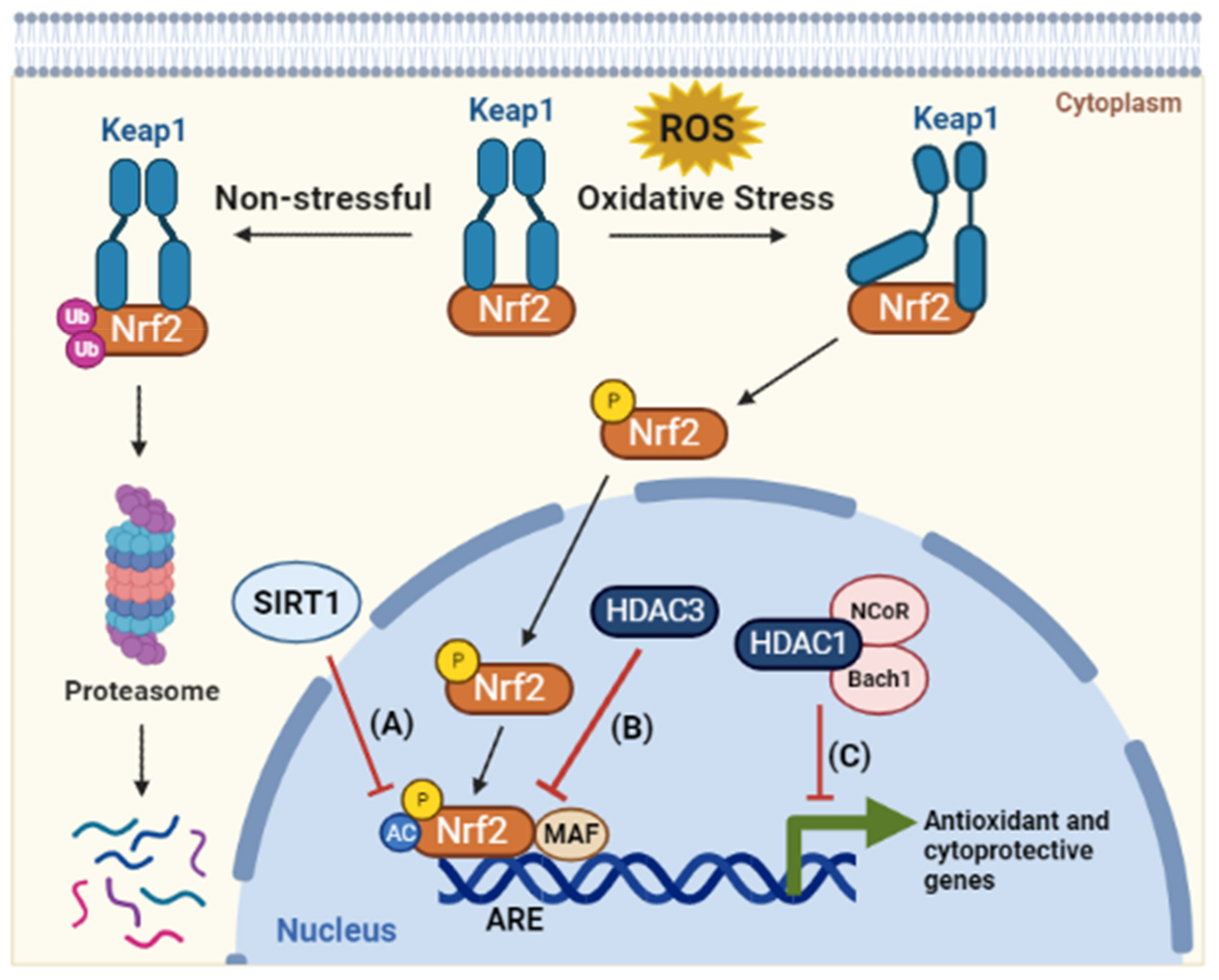
2.3. Oxidative Stress
2.4. Metabolic Stress (Hypoglycemia and Hypoxia)
References
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63.
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100.
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangi-ocarcinoma. Cells 2020, 9, 780.
- Li, Y.; Wang, F.; Chen, X.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent Deacetylase (HDAC) Inhibitors with Different Zinc Binding Groups. Curr. Top. Med. Chem. 2019, 19, 223–241.
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and Disease: The Road Ahead. Front. Pharmacol. 2012, 3, 4.
- Chiaradonna, F.; Cirulli, C.; Palorini, R.; Votta, G.; Alberghina, L. New Insights into the Connection between Histone Deacetylases, Cell Metabolism, and Cancer. Antioxid. Redox Signal. 2015, 23, 30–50.
- Barneda-Zahonero, B.; Parra, M. Histone Deacetylases and Cancer. Mol. Oncol. 2012, 6, 579–589.
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831.
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90: A Novel Basis for Antileukemia Activity of Histone Deacetylase Inhibitors. J. Biol. Chem. 2005, 280, 26729–26734.
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The Cytoplasmic Deacetylase HDAC6 Is Required for Efficient Oncogenic Tumorigenesis. Cancer Res. 2008, 68, 7561–7569.
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626.
- Deakin, N.O.; Turner, C.E. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol. 2014, 206, 395–413.
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394.
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189.
- Zeng, L.-S.; Yang, X.-Z.; Wen, Y.-F.; Mai, S.-J.; Wang, M.-H.; Zhang, M.-Y.; Zheng, X.S.; Wang, H.-Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Ageing 2016, 8, 1236–1248.
- Kang, Z.-H.; Wang, C.-Y.; Zhang, W.-L.; Zhang, J.-T.; Yuan, C.-H.; Zhao, P.-W.; Lin, Y.-Y.; Hong, S.; Li, C.-Y.; Wang, L. Histone Deacetylase HDAC4 Promotes Gastric Cancer SGC-7901 Cells Progression via p21 Repression. PLoS ONE 2014, 9, e98894.
- Hagelkruys, A.; Sawicka, A.; Rennmayr, M.; Seiser, C. The Biology of HDAC in Cancer: The Nuclear and Epigenetic Components. Handb. Exp. Pharmacol. 2011, 206, 13–37.
- Ropero, S.; Esteller, M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25.
- Minucci, S.; Nervi, C.; lo Coco, F.; Pelicci, P.G. Histone Deacetylases: A Common Molecular Target for Differentiation Treat-ment of Acute Myeloid Leukemias? Oncogene 2001, 20, 3110–3115.
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor α. Int. J. Cancer 2003, 107, 353–358.
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463.
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-I.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968.
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32.
- Alhmoud, J.F.; Woolley, J.F.; al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050.
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087.
- Lavin, M.F.; Shiloh, Y. The Genetic Defect in Ataxia-Telangiectasia. Annu. Rev. Immunol. 1997, 15, 177–202.
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027.
- Tsukamoto, Y.; Kato, J.; Ikeda, H. Silencing Factors Participate in DNA Repair and Recombination in Saccharomyces Cerevisiae . Nature 1997, 388, 900–903.
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153.
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 2008, 4, e1000155.
- Zhong, J.; Ji, L.; Chen, H.; Li, X.; Zhang, J.; Wang, X.; Wu, W.; Xu, Y.; Huang, F.; Cai, W.; et al. Acetylation of hMOF Modulates H4K16ac to Regulate DNA Repair Genes in Response to Oxidative Stress. Int. J. Biol. Sci. 2017, 13, 923–934.
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031.
- Orlando, G.; Khoronenkova, S.V.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. ARF induction in response to DNA strand breaks is regulated by PARP1. Nucleic Acids Res. 2013, 42, 2320–2329.
- Stengel, K.R.; Hiebert, S.W. Class I HDACs Affect DNA Replication, Repair, and Chromatin Structure: Implications for Cancer Therapy. Antioxid. Redox Signal. 2015, 23, 51–65.
- Alabert, C.; Groth, A. Chromatin Replication and Epigenome Maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167.
- Wells, C.E.; Bhaskara, S.R.; Stengel, K.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of Histone Deacetylase 3 Causes Replication Stress in Cutaneous T Cell Lymphoma. PLoS ONE 2013, 8, e68915.
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.-W.; Hiebert, S.W. Deletion of Histone Deacetylase 3 Reveals Critical Roles in S Phase Progression and DNA Damage Control. Mol. Cell 2008, 30, 61–72.
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262.
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738.
- Gokhale, S.; Nyayanit, D.; Gadgil, C. A systems view of the protein expression process. Syst. Synth. Biol. 2011, 5, 139–150.
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. HDAC Inhibitors and RECK Modulate Endoplasmic Reticulum Stress in Tumor Cells. Int. J. Mol. Sci. 2017, 18, 258.
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607.
- Sakamoto, K.M.; Aldana-Masangkay, G.I. The Role of HDAC6 in Cancer. J. Biomed. Biotechnol. 2011, 2011, 875824.
- Krämer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6–HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509.
- Lin, T.-Y.; Fenger, J.; Murahari, S.; Bear, M.D.; Kulp, S.K.; Wang, D.; Chen, C.-S.; Kisseberth, W.C.; London, C.A. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood 2010, 115, 4217–4225.
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.-P.; Vourc’H, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181.
- Ahn, M.Y.; Kang, D.O.; Na, Y.J.; Yoon, S.; Choi, W.S.; Kang, K.W.; Chung, H.Y.; Jung, J.H.; Min, D.S.; Kim, H.S. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett. 2012, 325, 189–199.
- Jeon, H.W.; Lee, Y.M. Inhibition of Histone Deacetylase Attenuates Hypoxia-Induced Migration and Invasion of Cancer Cells via the Restoration of RECK Expression. Mol. Cancer Ther. 2010, 9, 1361–1370.
- Takahashi, C.; Sheng, Z.; Horan, T.P.; Kitayama, H.; Maki, M.; Hitomi, K.; Kitaura, Y.; Takai, S.; Sasahara, R.M.; Horimoto, A.; et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc. Natl. Acad. Sci. USA 1998, 95, 13221–13226.
- Chen, Y.; Tseng, S.-H. The potential of RECK inducers as antitumor agents for glioma. Anticancer Res. 2012, 32, 2991–2998.
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443.
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821.
- Chen, M.; Xie, S. Therapeutic targeting of cellular stress responses in cancer. Thorac. Cancer 2018, 9, 1575–1582.
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Targeting Oxidative Stress in Cancer. Expert Opin. Ther. Targets 2010, 14, 1225–1245.
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86.
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Biol. Chem. 2011, 286, 7629–7640.
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 713–727.
- Dohi, Y.; Ikura, T.; Hoshikawa, Y.; Katoh, Y.; Ota, K.; Nakanome, A.; Muto, A.; Omura, S.; Ohta, T.; Ito, A.; et al. Bach1 inhibits oxidative stress–induced cellular senescence by impeding p53 function on chromatin. Nat. Struct. Mol. Biol. 2008, 15, 1246–1254.
- Kim, S.-H.; Jeong, J.-W.; Park, J.A.; Lee, J.-W.; Seo, J.H.; Jung, B.-K.; Bae, M.-K.; Kim, K.-W. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651.
- Yoo, Y.-G.; Kong, G.; Lee, M.-O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1α protein by recruiting histone deacetylase 1. EMBO J. 2006, 25, 1231–1241.
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone Deacetylase 7 Associates with Hypoxia-inducible Factor 1α and Increases Transcriptional Activity. J. Biol. Chem. 2004, 279, 41966–41974.
- Hiraoka, N.; Allen, E.; Apel, I.J.; Gyetko, M.R.; Weiss, S.J. Matrix Metalloproteinases Regulate Neovascularization by Acting as Pericellular Fibrinolysins. Cell 1998, 95, 365–377.
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658.
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293.
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.; Cardoso, S.M.; Clish, C.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428.
- White, E. Role of the Metabolic Stress Responses of Apoptosis and Autophagy in Tumor Suppression. In Oncogenes Meet Metabolism; Springer: Berlin/Heidelberg, Germany, 2007; pp. 23–24.
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200.
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Cancer 2016, 16, 613–614.
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264.
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689.
- Bhardwaj, A.; Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547.
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.K.A.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696.
- Greene, K.S.; Lukey, M.J.; Wang, X.; Blank, B.; Druso, J.E.; Lin, M.-C.J.; Stalnecker, C.A.; Zhang, C.; Abril, Y.N.; Erickson, J.W.; et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 26625–26632.
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9.
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.K.; et al. SIRT 2-dependent IDH 1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, 21, e48183.
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693.
- Wang, W.; Zeng, Y.; Wu, B.; Deiters, A.; Liu, W.R. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem. Biol. 2016, 11, 1973–1981.
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492.
- Kutil, Z.; Mikešová, J.; Zessin, M.; Meleshin, M.; Nováková, Z.; Alquicer, G.; Kozikowski, A.; Sippl, W.; Bařinka, C.; Schutkowski, M. Continuous Activity Assay for HDAC11 Enabling Reevaluation of HDAC Inhibitors. ACS Omega 2019, 4, 19895–19904.

