Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Eleni Manthou | -- | 3200 | 2022-08-03 08:18:12 | | | |
| 2 | Sirius Huang | Meta information modification | 3200 | 2022-08-04 06:04:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Miliaras, D.; Kesidou, E.; Boziki, M.; Petratos, S.; Grigoriadis, N.; Theotokis, P. Molecular Signals Driving Myelinogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/25790 (accessed on 27 July 2026).
Dermitzakis I, Manthou ME, Meditskou S, Miliaras D, Kesidou E, Boziki M, et al. Molecular Signals Driving Myelinogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/25790. Accessed July 27, 2026.
Dermitzakis, Iasonas, Maria Eleni Manthou, Soultana Meditskou, Dimosthenis Miliaras, Evangelia Kesidou, Marina Boziki, Steven Petratos, Nikolaos Grigoriadis, Paschalis Theotokis. "Molecular Signals Driving Myelinogenesis" Encyclopedia, https://encyclopedia.pub/entry/25790 (accessed July 27, 2026).
Dermitzakis, I., Manthou, M.E., Meditskou, S., Miliaras, D., Kesidou, E., Boziki, M., Petratos, S., Grigoriadis, N., & Theotokis, P. (2022, August 03). Molecular Signals Driving Myelinogenesis. In Encyclopedia. https://encyclopedia.pub/entry/25790
Dermitzakis, Iasonas, et al. "Molecular Signals Driving Myelinogenesis." Encyclopedia. Web. 03 August, 2022.
Copy Citation
As the regulator of all cognitive, sensory, and motor activity, the nervous system is the most complex biological system in humans. In order for myelinogenesis to happen, neural stem cells (NSCs) need to undergo specific developmental stages, with the process of oligodendrogenesis, as well as additional steps for the maintenance of these primary myelin sheaths.
oligodendrogenesis
myelinogenesis
myelin formation
embryology
CNS development
1. Introduction
As has been articulated from the experimental evidence, the inauguration of myelinogenesis necessitates the formation of OPCs from multipotent NSCs, which ultimately give rise to mature myelinating OLs through a multistep process (Figure 1) [1][2]. A vital step herein lies in OPCs’ ability to migrate toward miscellaneous sites and proliferate, based predominately on environmental stimuli. These cells become post-mitotic, exiting the cell cycle to express a substantial amount of myelin-associated proteins and differentiate into mature pre-myelinating OLs [3]. Following the proper recognition, targeting and ensheathing specific nerve fibers is the subsequent critical milestone where each pioneer process creates lamellar extensions that stretch and elaborate circumferentially around the target axon [2]. As a new membrane is generated at the leading edge of the forming myelin sheath’s inner tongue, which starts to resemble a spiral cross-sectional shape, the sheath continues to spread along the axonal length. The secured stability and maintenance of a newly-formed myelin sheath is the concluding event.
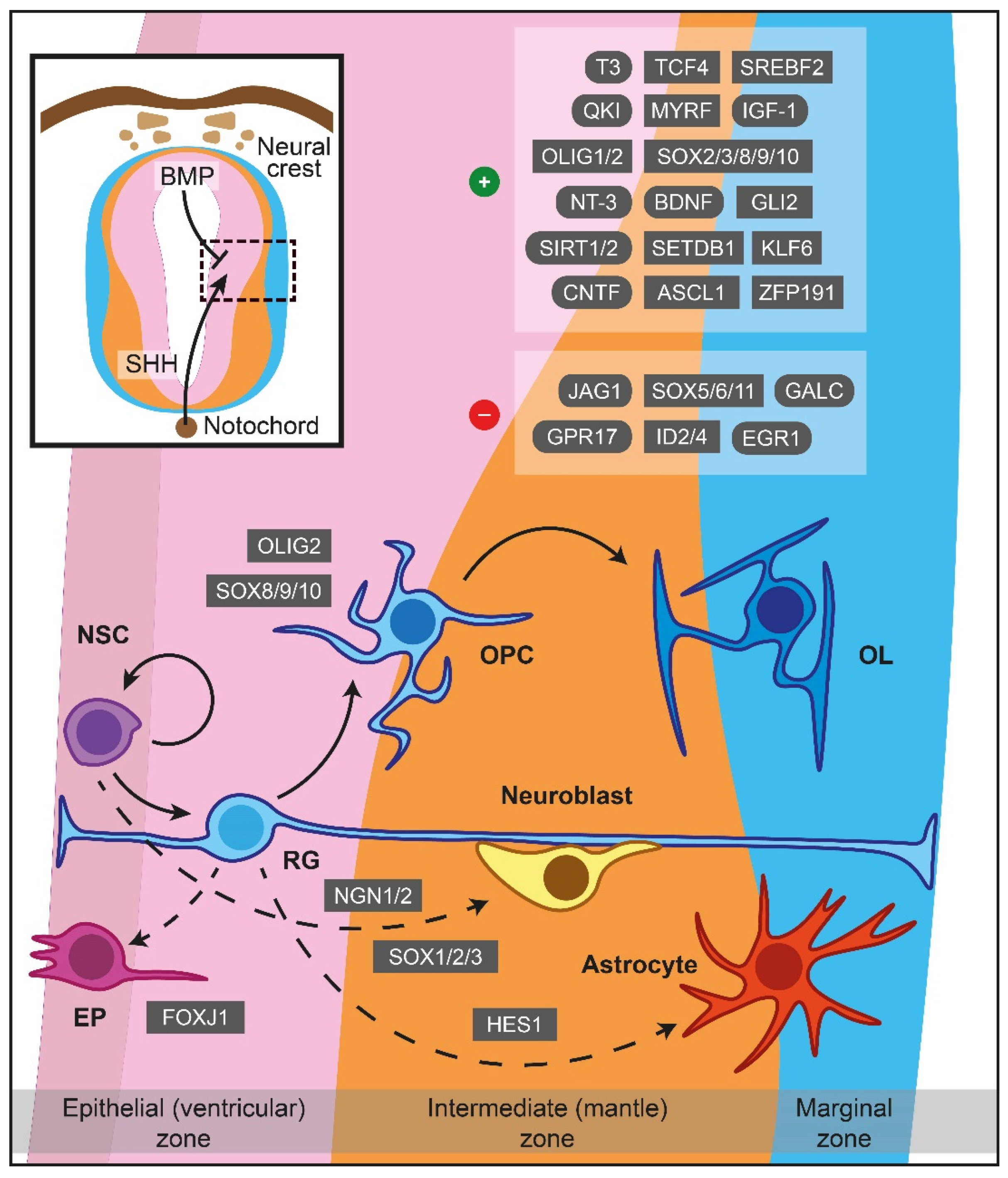
Figure 1. Major cues of OPCs generation and differentiation during myelinogenesis in the prenatal period. In the neuroepithelium lining the neural tube, NSCs are under the influence of notochord-derived SHH, which drives the cells to become OPCs through OLIG2, SOX8/9/10 or follow neuronal fate (neuroblasts) via NGN1/2 and SOX1/2/3. BMP originated from the neural crest instructing NSCs to become astrocytes, controlled by HES1 as well. FOXJ1 is a crucial transcription factor for the ependymal trajectory. The positive and negative cues controlling OPCs differentiation are displayed in the upper right boxes. The hatched box depicts a representative area around sulcus limitans (between alar and basal plates). Dashed lines showcase naturally occurring processes, albeit not addressed in detail herein. ASCL1: Achaete-scute family bHLH transcription factor 1, BDNF: Brain-derived neurotrophic factor, BMP: Bone morphogenetic protein, CNTF: Ciliary neurotrophic factor, EGR1: Early growth response 1, EP: Ependymal cells, FOXJ1: Transcription factor forkhead box J1, GALC: Galactosylceramidase, GLI2: Glioma-associated oncogene family zinc finger 2, GPR17: G protein-coupled receptor 17, HES1: Hes family bHLH transcription factor 1, ID2: Inhibitor of DNA binding 2, ID4: Inhibitor of DNA binding 2, IGF-1: Insulin-like growth factor 1, JAG1: Jagged canonical Notch ligand 1, KLF6: Kruppel-like factor 6, MYRF: Myelin regulatory factor, NGN1: Neurogenin-1, NGN2: Neurogenin-2, NSC: Neural stem cells, NT-3: Neurotrophin 3, OL: Oligodendrocytes, OLIG1: Oligodendrocyte transcription factor 1, OLIG2: Oligodendrocyte transcription factor 2, OPC: Oligodendrocyte precursor cell, QKI: Quaking homolog, KH domain RNA binding, RG: Radial glia, SET domain bifurcated histone lysine methyltransferase 1, SETDB1: SHH: Sonic hedgehog signaling molecule, SIRT1: Sirtuin 1, SIRT2: Sirtuin 2, SOX: Sex-determining region Y-box transcription factor, SREBF2: Sterol regulatory element-binding transcription factor 2, T3: Triiodothyronine, TCF4: Transcription factor 4, ZFP191: Zinc finger protein 191.
2. Formation of OPCs
OPCs being generated from the ventral VZ are under the influence of the morphogen molecule Sonic hedgehog (SHH) secreted from the notochord, while the dorsal counterparts are SHH-independent [4][5]. SHH signalling drives NSCs into a neuronal or OLs lineage fate superseding the effect of bone morphogenetic proteins (BMPs) which favour astroglial generation (Figure 1) [6][7]. Early secretion of SHH promotes motor neuron lineage formation, while interaction in later time periods promotes OLs differentiation [8]. Interestingly, the concentration of SHH can be controlled by sulfatase 1 expression in the ventral neuroepithelium prior to OPCs specification [9], whereas fibroblast growth factor (FGF) signalling is of paramount importance for further OLs differentiation, especially in the spinal cord [10][11].
Oligodendrocyte transcription factor 2 (OLIG2) is the primary regulator of OPCs generation [12][13], and its gene expression can be potentially repressed throughout the pre to postnatal period by paired box 6 (PAX6), Brahma-related gene-1 (BRG1), Iroquois homeobox 3 (IRX3), histone deacetylase (HDAC) 1, HDAC2, Distal-less homeobox (DLX) 1 and DLX 2 [14][15][16][17][18][19][20]. On the other hand, oligodendrocyte transcription factor 1 (OLIG1) is activated in later stages of OLs development [21]. Interestingly, the Hes family bHLH transcription factor (HES1) can drive RG to an astrocytic phenotype [22], while co-occurrence of OLIG2 with neurogenin-1 or neurogenin-2 supports motor neuron production [17][23][24].
Members of the sex-determining region Y-box transcription factor (SOX) family, such as SOX1, SOX2, and SOX3, can also direct OPCs towards a neuronal fate [12], in contrast to SOX8, SOX9, and SOX10, which favour the turnover of NSCs to OPCs in an autonomous manner [13][14][15]. Additionally, transcription factor forkhead box J1 (FOXJ1) supports the retention of RGs as ependymal cells throughout ventricles. Lastly, glioma-associated oncogene family zinc finger 2 (GLI2), myelin transcription factor 1 (MYT1), NK2 homeobox 6 (NKX2-6), and chromodomain-helicase-DNA-binding protein 8 (CHD8), among others, are embryonic cues for OLs specification that vary within CNS regions indicating brain region specificity [16][17][18][19].
3. Migration
SHH presence is equally catalytic to OPCs migration [20]. Platelet-derived growth factor subunit A (PDGFA) and its cognate receptor, PDGF receptor alpha (PDGFRα), are essential positive drivers for OPCs migration [21]. In line with this, SOX5, SOX6, SOX9, and SOX10 stimulate the migration, ensuring PDGF responsiveness [22][23]. Chondroitin sulfate proteoglycan neuron-glia antigen 2 (NG2) and ephrin-B2/B3 molecules control OPCs polarity and contact abilities, promoting or intercepting migration, respectively [24][25]. Nestin, neural cell adhesion molecule (NCAM), and OLIG1 can also act as chemoattractants, determining cytoskeletal plasticity as well as OPCs motility [26][27][28][29][30]. Other migration chemoattractants are 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNPase), OLIG2, hepatocyte growth factor (HGF), thrombospondin 1, endothelin 1 (ET-1), oligodendrocyte specific protein (OSP), OSP–associated protein (OAP-1), N-cadherin (NCAD), merosin, fibronectin, and integrin subunit beta 1 (αvβ1 integrin) [31][32][33][34][35][36][37][38][39]. Spassky et al. suggested that netrin-1 is a candidate mediator for chemoattraction during migration [40]. However, other studies considered this molecule as a chemorepellent, antagonizing PDGF [41][42].
More growth factors and associated molecules, such as vascular endothelial growth factor A (VEGF-A) combined with VEFG receptor 2 (VEGFR2), can act as chemoattractant molecules for OPCs migration along with miscellaneous members of the transforming growth factor beta (TGF-β) family (e.g., BMP7 and BMP4), and Gαi-linked sphingosine-1-phosphate receptor (S1PR) 1 and S1PR3 [43][44][45]. In contrast to these specific sphingosine molecules, S1PR2 and S1PR5 negatively regulated migration [45]. Moreover, although C-X-C motif chemokine receptor (CXCR) 4, C-X-C motif chemokine ligand (CXCL) 12 and semaphorin 3F have chemoattractive effects on the OPCs migration, semaphorin 3A, CXCL1, and CXCR2 inhibit migration [40][46][47]. In addition, tenascin-c inhibited OPCs migration, whilst both claudin (CLDN) 1 and CLDN3 supported OPCs relocation, validated also in human specimens [38][48][49].
4. Proliferation
Specific driver molecules that participate in migration, such as PDGFA and PDGFRα, contribute additionally to the OPC proliferation [26][50]. Interestingly, in the spinal cord, the mitogenic effect of PDGF was enhanced by chemokine CXCL1 and CXCR2 [23][51], while CXCL12 had a proliferative effect on OPCs, mediated by its receptor CXCR4 [52]. More growth factors, such as FGF2, brain-derived neurotrophic factor (BDNF), and epidermal growth factor (EGF) are shown to play a vital role in OPCs proliferation [53][54][55].
Associate developmental pathways are also implicated in this step; PDGF-mediated proliferation depends largely on Wnt/β-catenin and PI3K/AKT/mTOR pathways [56][57]. Furthermore, jagged canonical Notch ligand 1 (JAG1) promotes OPCs proliferation and critically blocks the subsequent differentiation step [58]. Carrying on subcellular, CHD7 and CHD8 regulate gene expression in specific brain regions [59][60]. Another member of the SOX family, SOX9, supports the development of OLs in the cerebellum, regulating the timing of proliferation [61]. MYT1, NCAM, cyclin-dependent kinase inhibitor 1B (p27KIP1), oligodendrocyte myelin glycoprotein (OMgp), and tubulin polymerization promoting protein (TPPP) are negative regulator cues for OPCs proliferation [62][63][64][65][66]. Interestingly, overexpression of inhibitor of DNA binding (ID) 2 and ID4 enhances proliferation [67][68]. Similarly, expression of SHH, HGF, neurotrophin-4 (NT-4), noggin, superoxide dismutase 1 (SOD1), neurotrophin-3 (NT-3), achaete-scute family bHLH transcription factor 1 (ASCL1), PAX6, CLDN1, and CLDN3 promotes the proliferation process [20][33][49][69][70][71][72][73][74].
Integrin-mediated signalling and, more specifically, OSP, OAP-1, αvβ1 integrin, αvβ3 integrin, fibronectin and laminin are pivotal mediators in cytoskeletal remodelling of proliferating OPCs [35][75][76]. Gadea et al. revealed that ET-1 is a candidate molecule for enhancing cell migration without influencing proliferation [39]. Later, Adams and colleagues underscored that loss of ET-1 reduces OPCs proliferation in the developing SVZ via directly binding to endothelin type B receptor (ETBR) [77]. A reduced OPCs proliferation is observed in GS homeobox 1/2 (Gsx1/2) mutant embryos, whereas galectin-4 (GAL-4) treatment increased the proliferation [78][79]. At last, NRG1 and SOX2 induce cell division [80][81]; however, the latest data demonstrate that NRG1 acting via ErbB did not alter the proliferation state of OPCs [82].
5. Differentiation
OLIG1 and OLIG2 are heavily involved in the post-proliferating step of myelinogenesis, defining the initiation of OPCs differentiation (Figure 1) [30][32][83], while BMPs seem to inhibit this process by downregulating myelin protein expression [84]. The effect can be reversed by using a physiological antagonist of BMP4, such as noggin, which may restore differentiation [70][85][86]. OLIG2 appears to interact with a variety of factors, such as ASCL1, BRG1, transcription factor 4 (TCF4), and SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) to ensure proper OPCs differentiation [87][88][89][90][91]. G protein-coupled receptor 17 (GPR17) can act as a downregulator of OLIG1 that negatively controls the maturation and coordinates the generation of myelinating OLs from pre-myelinating OLs through ID proteins [92]. Although overexpression of ID2 and ID4 both regulate myelin gene expression by inhibiting OLs differentiation [68][93], they are not the major in vivo repressors of differentiation [94]. Moreover, decreased levels of OLIG1 and myelin regulatory factor (MYRF) were observed under early growth response 1 (EGR1) and SOX11 overexpression, delineating the inhibitory action of the latest in OPCs differentiation [95][96]. Intriguingly, MYRF is a unique regulator participating in the late stages of OLs maturation and myelination, while the action of the other OLs’ lineage transcription factors is restricted on OPCs specification or initial differentiation of OLs [97].
SOX family proteins are also participating in the OLs differentiation. In particular, SOX2 and SOX3, through negative regulation of miR145, promote OLs maturation [98], while SOX5 and SOX6 increase PDGFRα expression, maintaining OLs in their immature state [23]. For the terminal differentiation of OLs, SOX8, SOX9, and SOX10 are required [61][99][100][101]. The state of myelinogenesis-associated gene expression is uniformly affected by NKX2-2 and NKX2-6 [19][102][103]. Ji et al. suggested a mechanism regarding NKX2-2-mediated inhibition of OLs differentiation via regulation of sirtuin 2 (SIRT2), which generally is a positive cue for OLs maturation [104]. Similarly, sirtuin 1 (SIRT1) participates in the differentiation of OPCs during development [105] through cytoskeleton-related OLs proteins. The Kruppel-like factor 6 (KLF6) is another transcription factor promoting OPCs differentiation through glycoprotein 130 (GP130)-signal transducer and transcription activator 3 (STAT3) signalling [106]. Growth factor-wise, BDNF is a regulator of OLs differentiation operating via binding to tyrosine receptor kinase B (TrkB) and enhancing the MAPK pathway to upregulate gene expression during OLs maturation [54][56][107]. Evidently, NT-3 is important for the transition of immature OLs to myelin-forming cells by recruiting c-Fos protein-activating protein kinase C (PKC) and tyrosine kinase activities [108][109]. Insulin-like growth factor 1 (IGF-1) is another main factor in assisting the development of OPCs to mature OLs [110]. In accordance with that, GRB2 associated binding protein 1 (GAB1) absence decreased OLs differentiation, acting as a novel target of PDGF [111]. Incidentally, Canoll et al. suggested that NRG1 is a negative regulator of OPCs differentiation [80], while Brinkmann et al. later demonstrated that NRG1 is required for OPCs differentiation [82].
As far as metabolism is concerned, quaking homolog, KH domain RNA binding (QKI)-5 forms a complex with sterol regulatory element-binding transcription factor 2 (SREBF2) that regulates the transcription of genes responsible for cholesterol biosynthesis in OLs during differentiation [112]. Lack of transactive response DNA-binding protein 43 (TDP-43) results in lower SREBF2 and low-density lipoprotein receptor (LDLR) expression and cholesterol levels in vitro and in vivo, indicating the potential role of TDP-43 in cholesterol homeostasis in OLs, which is linked with the proper completion of OLs development [113]. In the same manner, ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6) participates in OLs maturation via a supplement of OLs with choline [114]. Most importantly, triiodothyronine (T3) is a key molecule for blocking OPCs proliferation and promoting their differentiation into mature OLs [115][116]. Thyroid hormone receptor alpha (TRα) is found both in OPCs and mature OLs, whilst thyroid hormone receptor isoform beta 1 (TRβ1) is located only in mature OLs [117]. The OPCs differentiation is mediated by the TRα, while TRβ1 is responsible for promoting myelinogenesis in later stages [56]. Overexpression of HES5 decreases the levels of TRβ1 receptors, while ASCL1 increases them, demonstrating their role in regulating OLs differentiation timing [118]. The neurogenic locus notch homolog protein 1 (NOTCH1) is another receptor that also regulates the differentiation timing [119]. Interestingly, JAG1 is a receptor’s ligand responsible for inhibiting OLs differentiation, while contactin 1 (CNTN1) is another ligand with the opposite function [58][120].
Other membrane molecules which repress OPCs differentiation are NCAM and leucine-rich repeat, and Ig-like domain-containing Nogo receptor interacting protein 1 (LINGO-1) [121][122]. OLs maturation is negatively affected by GAL-4 and galactosylceramidase (GALC), while prominin-1, GLI2, p21-activated kinase 1 (PAK1), myelin-associated glycoprotein (MAG), SOD1, ciliary neurotrophic factor (CNTF), and inward rectifying potassium channel 4.1 (Kir4.1) are crucial for proper differentiation [17][74][79][123][124][125][126][127][128]. On the other hand, proper completion of OLs differentiation requires zinc finger protein 191 (ZFP191) [129]. Microtubule-associated protein 2 (MAP2), microtubule-associated protein tau (MAPT), CNPase, and TPPP may be involved in OLs differentiation by organising the microtubule system, similar to fasciculation and elongation protein zeta 1 (FEZ1), which is responsible for developing OLs processes’ arbour [66][130][131][132]. Additionally, important molecules being involved in the completion of OLs development are OMgp, brain enriched myelin-associated protein 1 (BCAS1) and glutathione (GSH) [133][134][135][136]. Myelin proteolipid protein (PLP) and myelin basic protein (MBP) are the main myelin structural proteins, but it is suggested that they play an additional role in OLs differentiation [137][138]. CLDN1 and CLDN3 control MBP, OLIG2, PLP, and SOX10 expression: these molecules are essential for OLs differentiation, indicating that claudins are needed [49]. Finally, connexin 47 (CX47) and adenosine triphosphate binding cassette subfamily D member 1 (ABCD1) may support OLs during their differentiation, aiding in gap junction coupling and reducing oxidative stress, respectively [139][140][141][142][143].
6. Ensheathment
Multiple positive cues are important for the inauguration of ensheathment (Figure 2). Amongst the prime ones with a positive effect on axon-glial junction maintenance is NCAD, which regulates the interaction between OLs processes and axons [144]. The L1 cell adhesion molecule (L1-CAM) and laminin expressed in axons bind to contactin and integrin located in OLs [145]. Upon the formation of the first loops/wraps, neurofascin 155 (NF155), located in paranodal loops, forms a well-defined complex with contactin-associated protein (CASPR) and CNTN1, transmembrane proteins which are expressed in axons [146][147][148]. The activation of this complex has a pivotal role in myelin targeting, sheath growth, organisation of paranodal loops and, therefore, supporting the axoglial junction [149][150]. However, CASPR does not participate in myelin targeting [149]. In juxtaparanodes, the axoglial junction is strengthened when transient axonal glycoprotein-1 (TAG-1), a crucial molecule for maintaining enrichment of Kv1.1/Kv1.2 channels [151], interacts with CASPR2. Regarding internodal axoglial adhesion, glial cell adhesion molecule (CADM) 4 binds to axonal CADM2 and CADM3, facilitating myelin targeting, axon wrapping, and myelin sheath growth [152]. Similarly, CADM1b strongly binds to axonal CADM2, positively regulating ensheathment and strengthening the junction [153]. In the same region of the myelin sheath, MAG binds to ganglioside in axons, especially ganglioside GD1a and GT1b, and enforces the junction’s stability [154][155].

Figure 2. Axoglial driving cues for the initiation of ensheathment during myelinogenesis. A process of oligodendrocyte (blue) approaches the axon (brown) based on their surfaces’ attractive and repulsive signals. The red-colored shapes represent negative surface molecules; the green ones stand for positive and the yellow for bidirectional signals. For illustrational purposes, the paranode, juxtaparanode, and internode regions are simplified. CADM1b: Cell adhesion molecule 1b, CADM2: Cell adhesion molecule 2, CADM3: Cell adhesion molecule 3, CADM4: Cell adhesion molecule 4, CASPR: Contactin-associated protein, CASPR2: Contactin-associated protein-like 2, CNTN1: Contactin 1, EphA4: Ephrin receptor A4, EphB: Ephrin receptor B, EphB1: Ephrin receptor B1, GAL-4: Galectin-4, GD1a: Ganglioside GD1a, GT1b: Ganglioside GT1b, L1-CAM: L1 cell adhesion molecule, LINGO-1: Leucine-rich repeat and Ig-like domain-containing Nogo receptor interacting protein 1, MAG: Myelin-associated glycoprotein, NCAD: N-cadherin, NCAM: Neural cell adhesion molecule, NF155: Neurofascin 155, TAG-1: Transient axonal glycoprotein-1.
Based on several studies, ephrins (A, B) and cognate receptors (A, B) have dual roles that rely on location and expression. While ephrin receptor (Eph) A4 in OLs is activated by axonal ephrin-A1 ligand, which inhibits the stability of axoglial junctions needed for ensheathment, EphA4, expressed in the axon surface, interacts with ephrin-B, promoting myelin sheet formation [156][157]. In addition, EphB1 of axons is activated through ephrin-B in OLs, which in turn stimulates myelinogenesis [157]. The axonal ephrinB2 via binding with EphB OLs receptor influences integrin activation, reducing myelin sheet formation [157]. The list of negative cues includes LINGO-1, which is located in both axons and OLs, and self-interacts in trans to control the number of targeted axons inhibiting myelinogenesis [122][158]. The NCAM is a cell adhesion molecule negatively regulating myelinogenesis. The downregulation of this protein is essential for promoting myelin formation during development, as myelinogenesis occurs only on NCAM negative axons [159]. A somatodendritic protein, junctional adhesion molecule 2 (JAM2) inhibits oligodendroglial interaction, suppressing myelinogenesis [160]. Apart from the somatodendritic molecules, GAL-4 is expressed only to unmyelinated segments of neurons in hippocampal and cortical regions; this protein is demonstrated as the first identified inhibitor of myelinogenesis in axons [161]. Of particular interest is the possible role of OLIG1 in axonal recognition during myelinogenesis [162].
7. Myelin Sheath Growth and Preservation
The long-term membrane expansion and maintenance of the newly-formed myelin sheath is the final step in completing myelinogenesis and is utterly controlled by the major myelin proteins. The most abundant myelin proteins are PLP (>50%) and MBP (~15%), having a significant role in the stabilization of the myelin structure [2][31][163]. The disruption of PLP gene expression presents impaired membrane compaction [164]. MAG, on the other side, is the third most abundant protein in CNS myelin (~5%), and does not seem to contribute to maintenance as much as it does to the previously described initial interaction between OLs and axons [126][165]. Interestingly, myelin oligodendrocyte glycoprotein (MOG) [166][167], CNPase [31][168], myelin-associated oligodendrocyte basic protein (MOBP) [169], and OMgp [65][170][171], all minor CNS myelin proteins (<1%), need more investigation on how they influence the formation and maintenance of myelin sheaths in compact myelin.
OLs microtubule stability is mediated by MAP2 and MAPT [130], while CX32 and CX47 participate in maintenance [140]. Claudins, such as OSP, CLDN1, and CLDN3, play a pivotal role as well [49][164]. Transcription factors that participate in the lamellar extension process are SOX8, SOX10, NKX2-2, NKX6-2, and MYRF [14][101][172][173]. Transmembrane protein (TMEM) 98, which inhibits the self-cleavage of MYRF, ID4, and OLIG1, could also be involved in the process [93][174], whereas OLIG2 is expressed only until myelin membranes’ production is completed [162][175]. In addition, the ERK1/2 MAP kinase pathway is indispensable in maintaining myelinated axons via FGF–FGF receptor 1 and 2 (FGFR1 and FGFR2) [176][177]. Experiments in Hdac3-mutant optic nerves raised the possibility that HDAC3 is also necessary for myelin integrity [178].
Proper cholesterol biosynthesis is prioritized in myelinogenesis, with QKI regulating this cholesterol production via SREBF2. Specifically, QKI-5 acts synergistically with peroxisome proliferator-activated receptor beta (PPARβ)-retinoid X receptor alpha (RXRα) activating transcription of the response in fatty acid metabolism genes. This operation of QKI-5 is significant for maintaining myelin homeostasis [112]. The ceramide galactosyl transferase (CGT) is a key enzyme for catalyzing GALC synthesis, while ceramide sulfotransferase (CST) is responsible for converting GALC to sulfatide [179][180]. Both CST and CGT mutant animals showed a regionally specific loss of myelin stability [179]. Thus, GALC and sulfatide have a pivotal role in the long-term maintenance of myelin, with the GALC being more crucial for myelin development than its assembly [179][180]. Additionally, peroxisomal metabolism also influences myelin survival [181]. For example, a peroxisomal transmembrane protein responsible for very long-chain fatty metabolism is encoded by the ABCD1 gene and is key in maintaining myelin stability [143][182]. Lastly, the age-dependent changes of TMEM10 might be linked with its action in maintaining CNS myelin [183].
References
- Grabel, L. Developmental Origin of Neural Stem Cells: The Glial Cell That Could. Stem Cell Rev. Rep. 2012, 8, 577–585.
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927.
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 37–53.
- Tripathi, R.B.; Clarke, L.E.; Burzomato, V.; Kessaris, N.; Anderson, P.N.; Attwell, D.; Richardson, W.D. Dorsally and Ventrally Derived Oligodendrocytes Have Similar Electrical Properties but Myelinate Preferred Tracts. J. Neurosci. 2011, 31, 6809–6819.
- Cai, J.; Qi, Y.; Hu, X.; Tan, M.; Liu, Z.; Zhang, J.; Li, Q.; Sander, M.; Qiu, M. Generation of Oligodendrocyte Precursor Cells from Mouse Dorsal Spinal Cord Independent of Nkx6 Regulation and Shh Signaling. Neuron 2005, 45, 41–53.
- Zhu, G.; Mehler, M.F.; Zhao, J.; Yu Yung, S.; Kessler, J.A. Sonic Hedgehog and BMP2 Exert Opposing Actions on Proliferation and Differentiation of Embryonic Neural Progenitor Cells. Dev. Biol. 1999, 215, 118–129.
- Gomes, W.A.; Mehler, M.F.; Kessler, J.A. Transgenic Overexpression of BMP4 Increases Astroglial and Decreases Oligodendroglial Lineage Commitment. Dev. Biol. 2003, 255, 164–177.
- Orentas, D.M.; Hayes, J.E.; Dyer, K.L.; Miller, R.H. Sonic Hedgehog Signaling Is Required during the Appearance of Spinal Cord Oligodendrocyte Precursors. Dev. Camb. Engl. 1999, 126, 2419–2429.
- Danesin, C.; Agius, E.; Escalas, N.; Ai, X.; Emerson, C.; Cochard, P.; Soula, C. Ventral Neural Progenitors Switch toward an Oligodendroglial Fate in Response to Increased Sonic Hedgehog (Shh) Activity: Involvement of Sulfatase 1 in Modulating Shh Signaling in the Ventral Spinal Cord. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5037–5048.
- Farreny, M.-A.; Agius, E.; Bel-Vialar, S.; Escalas, N.; Khouri-Farah, N.; Soukkarieh, C.; Danesin, C.; Pituello, F.; Cochard, P.; Soula, C. FGF Signaling Controls Shh-Dependent Oligodendroglial Fate Specification in the Ventral Spinal Cord. Neural Develop. 2018, 13, 3.
- Kearns, C.A.; Walker, M.; Ravanelli, A.M.; Scott, K.; Arzbecker, M.R.; Appel, B. Zebrafish Spinal Cord Oligodendrocyte Formation Requires Boc Function. Genetics 2021, 218, iyab082.
- Bylund, M.; Andersson, E.; Novitch, B.G.; Muhr, J. Vertebrate Neurogenesis Is Counteracted by Sox1–3 Activity. Nat. Neurosci. 2003, 6, 1162–1168.
- Stolt, C.C.; Schmitt, S.; Lommes, P.; Sock, E.; Wegner, M. Impact of Transcription Factor Sox8 on Oligodendrocyte Specification in the Mouse Embryonic Spinal Cord. Dev. Biol. 2005, 281, 309–317.
- Turnescu, T.; Arter, J.; Reiprich, S.; Tamm, E.R.; Waisman, A.; Wegner, M. Sox8 and Sox10 Jointly Maintain Myelin Gene Expression in Oligodendrocytes. Glia 2018, 66, 279–294.
- Pozniak, C.D.; Langseth, A.J.; Dijkgraaf, G.J.P.; Choe, Y.; Werb, Z.; Pleasure, S.J. Sox10 Directs Neural Stem Cells toward the Oligodendrocyte Lineage by Decreasing Suppressor of Fused Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 21795–21800.
- Wegner, M. Specification of Oligodendrocytes. In Patterning and Cell Type Specification in the Developing CNS and PNS; Elsevier: Amsterdam, The Netherlands, 2020; pp. 847–866. ISBN 978-0-12-814405-3.
- Qi, Y.; Tan, M.; Hui, C.-C.; Qiu, M. Gli2 Is Required for Normal Shh Signaling and Oligodendrocyte Development in the Spinal Cord. Mol. Cell. Neurosci. 2003, 23, 440–450.
- Hudson, L.D.; Romm, E.; Berndt, J.A.; Nielsen, J.A. A Tool for Examining the Role of the Zinc Finger Myelin Transcription Factor 1 (Myt1) in Neural Development: Myt1 Knock-in Mice. Transgenic Res. 2011, 20, 951–961.
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the Oligodendroglial Lineage, Myelination, and Remyelination. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2011, 1812, 184–193.
- Merchán, P.; Bribián, A.; Sánchez-Camacho, C.; Lezameta, M.; Bovolenta, P.; de Castro, F. Sonic Hedgehog Promotes the Migration and Proliferation of Optic Nerve Oligodendrocyte Precursors. Mol. Cell. Neurosci. 2007, 36, 355–368.
- Frost, E.E.; Zhou, Z.; Krasnesky, K.; Armstrong, R.C. Initiation of Oligodendrocyte Progenitor Cell Migration by a PDGF-A Activated Extracellular Regulated Kinase (ERK) Signaling Pathway. Neurochem. Res. 2009, 34, 169–181.
- Finzsch, M.; Stolt, C.C.; Lommes, P.; Wegner, M. Sox9 and Sox10 Influence Survival and Migration of Oligodendrocyte Precursors in the Spinal Cord by Regulating PDGF Receptor Aexpression. Development 2008, 135, 637–646.
- Baroti, T.; Zimmermann, Y.; Schillinger, A.; Liu, L.; Lommes, P.; Wegner, M.; Stolt, C.C. Transcription Factors Sox5 and Sox6 Exert Direct and Indirect Influences on Oligodendroglial Migration in Spinal Cord and Forebrain: SoxD Proteins in Oligodendrocyte Progenitors. Glia 2016, 64, 122–138.
- Biname, F.; Sakry, D.; Dimou, L.; Jolivel, V.; Trotter, J. NG2 Regulates Directional Migration of Oligodendrocyte Precursor Cells via Rho GTPases and Polarity Complex Proteins. J. Neurosci. 2013, 33, 10858–10874.
- Prestoz, L.; Chatzopoulou, E.; Lemkine, G.; Spassky, N.; Lebras, B.; Kagawa, T.; Ikenaka, K.; Zalc, B.; Thomas, J.-L. Control of Axonophilic Migration of Oligodendrocyte Precursor Cells by Eph–Ephrin Interaction. Neuron Glia Biol. 2004, 1, 73–83.
- Grinspan, J.B.; Franceschini, B. Platelet-Derived Growth Factor Is a Survival Factor for PSA-NCAM+ Oligodendrocyte Pre-Progenitor Cells. J. Neurosci. Res. 1995, 41, 540–551.
- Decker, L.; Avellana-Adalid, V.; Nait-Oumesmar, B.; Durbec, P.; Baron-Van Evercooren, A. Oligodendrocyte Precursor Migration and Differentiation: Combined Effects of PSA Residues, Growth Factors, and Substrates. Mol. Cell. Neurosci. 2000, 16, 422–439.
- Gallo, V.; Armstrong, R. Developmental and Growth Factor-Induced Regulation of Nestin in Oligodendrocyte Lineage Cells. J. Neurosci. 1995, 15, 394–406.
- Motizuki, M.; Isogaya, K.; Miyake, K.; Ikushima, H.; Kubota, T.; Miyazono, K.; Saitoh, M.; Miyazawa, K. Oligodendrocyte Transcription Factor 1 (Olig1) Is a Smad Cofactor Involved in Cell Motility Induced by Transforming Growth Factor-β. J. Biol. Chem. 2013, 288, 18911–18922.
- Zhou, Q.; Anderson, D.J. The BHLH Transcription Factors OLIG2 and OLIG1 Couple Neuronal and Glial Subtype Specification. Cell 2002, 109, 61–73.
- Yin, X.; Peterson, J.; Gravel, M.; Braun, P.E.; Trapp, B.D. CNP Overexpression Induces Aberrant Oligodendrocyte Membranes and Inhibits MBP Accumulation and Myelin Compaction. J. Neurosci. Res. 1997, 50, 238–247.
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of Olig2 Function in Oligodendrocyte Progenitors Promotes Remyelination. Brain 2015, 138, 120–135.
- Yan, H.; Rivkees, S.A. Hepatocyte Growth Factor Stimulates the Proliferation and Migration of Oligodendrocyte Precursor Cells. J. Neurosci. Res. 2002, 69, 597–606.
- Scott-Drew, S.; ffrench-Constant, C. Expression and Function of Thrombospondin-1 in Myelinating Glial Cells of the Central Nervous System. J. Neurosci. Res. 1997, 50, 202–214.
- Tiwari-Woodruff, S.K.; Buznikov, A.G.; Vu, T.Q.; Micevych, P.E.; Chen, K.; Kornblum, H.I.; Bronstein, J.M. OSP/Claudin-11 Forms a Complex with a Novel Member of the Tetraspanin Super Family and Beta1 Integrin and Regulates Proliferation and Migration of Oligodendrocytes. J. Cell Biol. 2001, 153, 295–305.
- Payne, H.R.; Hemperly, J.J.; Lemmon, V. N-Cadherin Expression and Function in Cultured Oligodendrocytes. Dev. Brain Res. 1996, 97, 9–15.
- Milner, R.; Edwards, G.; Streuli, C.; Ffrench-Constant, C. A Role in Migration for the Alpha V Beta 1 Integrin Expressed on Oligodendrocyte Precursors. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 7240–7252.
- Frost, E.; Kiernan, B.W.; Faissner, A.; ffrench-Constant, C. Regulation of Oligodendrocyte Precursor Migration by Extracellular Matrix: Evidence for Substrate-Specific Inhibition of Migration by Tenascin-C. Dev. Neurosci. 1996, 18, 266–273.
- Gadea, A.; Aguirre, A.; Haydar, T.F.; Gallo, V. Endothelin-1 Regulates Oligodendrocyte Development. J. Neurosci. 2009, 29, 10047–10062.
- Spassky, N.; de Castro, F.; Le Bras, B.; Heydon, K.; Quéraud-LeSaux, F.; Bloch-Gallego, E.; Chédotal, A.; Zalc, B.; Thomas, J.-L. Directional Guidance of Oligodendroglial Migration by Class 3 Semaphorins and Netrin-1. J. Neurosci. 2002, 22, 5992–6004.
- Jarjour, A.A.; Manitt, C.; Moore, S.W.; Thompson, K.M.; Yuh, S.-J.; Kennedy, T.E. Netrin-1 Is a Chemorepellent for Oligodendrocyte Precursor Cells in the Embryonic Spinal Cord. J. Neurosci. 2003, 23, 3735–3744.
- Tsai, H.-H.; Tessier-Lavigne, M.; Miller, R.H. Netrin 1 Mediates Spinal Cord Oligodendrocyte Precursor Dispersal. Development 2003, 130, 2095–2105.
- Zhang, H.; Vutskits, L.; Pepper, M.S.; Kiss, J.Z. VEGF Is a Chemoattractant for FGF-2–Stimulated Neural Progenitors. J. Cell Biol. 2003, 163, 1375–1384.
- Choe, Y.; Huynh, T.; Pleasure, S.J. Migration of Oligodendrocyte Progenitor Cells Is Controlled by Transforming Growth Factor Family Proteins during Corticogenesis. J. Neurosci. 2014, 34, 14973–14983.
- Novgorodov, A.S.; El-Awani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of Sphingosine-1-phosphate Receptor S1P5 Inhibits Oligodendrocyte Progenitor Migration. FASEB J. 2007, 21, 1503–1514.
- Tsai, H.-H.; Frost, E.; To, V.; Robinson, S.; Ffrench-Constant, C.; Geertman, R.; Ransohoff, R.M.; Miller, R.H. The Chemokine Receptor CXCR2 Controls Positioning of Oligodendrocyte Precursors in Developing Spinal Cord by Arresting Their Migration. Cell 2002, 110, 373–383.
- Dziembowska, M.; Tham, T.N.; Lau, P.; Vitry, S.; Lazarini, F.; Dubois-Dalcq, M. A Role for CXCR4 Signaling in Survival and Migration of Neural and Oligodendrocyte Precursors. Glia 2005, 50, 258–269.
- Garcion, E.; Faissner, A.; ffrench-Constant, C. Knockout Mice Reveal a Contribution of the Extracellular Matrix Molecule Tenascin-C to Neural Precursor Proliferation and Migration. Dev. Camb. Engl. 2001, 128, 2485–2496.
- Chen, Y.; Zheng, Z.; Mei, A.; Huang, H.; Lin, F. Claudin-1 and Claudin-3 as Molecular Regulators of Myelination in Leukoaraiosis Patients. Clinics 2021, 76, e2167.
- Calver, A.R.; Hall, A.C.; Yu, W.-P.; Walsh, F.S.; Heath, J.K.; Betsholtz, C.; Richardson, W.D. Oligodendrocyte Population Dynamics and the Role of PDGF In Vivo. Neuron 1998, 20, 869–882.
- Robinson, S.; Tani, M.; Strieter, R.M.; Ransohoff, R.M.; Miller, R.H. The Chemokine Growth-Regulated Oncogene-Alpha Promotes Spinal Cord Oligodendrocyte Precursor Proliferation. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 10457–10463.
- Kadi, L.; Selvaraju, R.; de Lys, P.; Proudfoot, A.E.I.; Wells, T.N.C.; Boschert, U. Differential Effects of Chemokines on Oligodendrocyte Precursor Proliferation and Myelin Formation in Vitro. J. Neuroimmunol. 2006, 174, 133–146.
- Frederick, T.J.; Min, J.; Altieri, S.C.; Mitchell, N.E.; Wood, T.L. Synergistic Induction of Cyclin D1 in Oligodendrocyte Progenitor Cells by IGF-I and FGF-2 Requires Differential Stimulation of Multiple Signaling Pathways. Glia 2007, 55, 1011–1022.
- Van’t Veer, A.; Du, Y.; Fischer, T.Z.; Boetig, D.R.; Wood, M.R.; Dreyfus, C.F. Brain-Derived Neurotrophic Factor Effects on Oligodendrocyte Progenitors of the Basal Forebrain Are Mediated through TrkB and the MAP Kinase Pathway. J. Neurosci. Res. 2009, 87, 69–78.
- Yang, J.; Cheng, X.; Qi, J.; Xie, B.; Zhao, X.; Zheng, K.; Zhang, Z.; Qiu, M. EGF Enhances Oligodendrogenesis from Glial Progenitor Cells. Front. Mol. Neurosci. 2017, 10, 106.
- Pukos, N.; Yoseph, R.; McTigue, D.M. To Be or Not to Be: Environmental Factors That Drive Myelin Formation during Development and after CNS Trauma. Neuroglia 2018, 1, 63–90.
- Hill, R.A.; Patel, K.D.; Medved, J.; Reiss, A.M.; Nishiyama, A. NG2 Cells in White Matter but Not Gray Matter Proliferate in Response to PDGF. J. Neurosci. 2013, 33, 14558–14566.
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch Receptor Activation Inhibits Oligodendrocyte Differentiation. Neuron 1998, 21, 63–75.
- Marie, C.; Clavairoly, A.; Frah, M.; Hmidan, H.; Yan, J.; Zhao, C.; Van Steenwinckel, J.; Daveau, R.; Zalc, B.; Hassan, B.; et al. Oligodendrocyte Precursor Survival and Differentiation Requires Chromatin Remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. USA 2018, 115, E8246–E8255.
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8.
- Hashimoto, R.; Hori, K.; Owa, T.; Miyashita, S.; Dewa, K.; Masuyama, N.; Sakai, K.; Hayase, Y.; Seto, Y.; Inoue, Y.U.; et al. Origins of Oligodendrocytes in the Cerebellum, Whose Development Is Controlled by the Transcription Factor, Sox9. Mech. Dev. 2016, 140, 25–40.
- Nielsen, J.A.; Berndt, J.A.; Hudson, L.D.; Armstrong, R.C. Myelin Transcription Factor 1 (Myt1) Modulates the Proliferation and Differentiation of Oligodendrocyte Lineage Cells. Mol. Cell. Neurosci. 2004, 25, 111–123.
- Amoureux, M.-C.; Cunningham, B.A.; Edelman, G.M.; Crossin, K.L. N-CAM Binding Inhibits the Proliferation of Hippocampal Progenitor Cells and Promotes Their Differentiation to a Neuronal Phenotype. J. Neurosci. 2000, 20, 3631–3640.
- Casaccia-Bonnefil, P.; Hardy, R.J.; Teng, K.K.; Levine, J.M.; Koff, A.; Chao, M.V. Loss of P27Kip1 Function Results in Increased Proliferative Capacity of Oligodendrocyte Progenitors but Unaltered Timing of Differentiation. Development 1999, 126, 4027–4037.
- Vourc’h, P.; Dessay, S.; Mbarek, O.; Marouillat Védrine, S.; Müh, J.-P.; Andres, C. The Oligodendrocyte-Myelin Glycoprotein Gene Is Highly Expressed during the Late Stages of Myelination in the Rat Central Nervous System. Brain Res. Dev. Brain Res. 2003, 144, 159–168.
- Lehotzky, A.; Lau, P.; Tokési, N.; Muja, N.; Hudson, L.D.; Ovádi, J. Tubulin Polymerization-Promoting Protein (TPPP/P25) Is Critical for Oligodendrocyte Differentiation. Glia 2010, 58, 157–168.
- Kondo, T. The Id4 HLH Protein and the Timing of Oligodendrocyte Differentiation. EMBO J. 2000, 19, 1998–2007.
- Wang, S.; Sdrulla, A.; Johnson, J.E.; Yokota, Y.; Barres, B.A. A Role for the Helix-Loop-Helix Protein Id2 in the Control of Oligodendrocyte Development. Neuron 2001, 29, 603–614.
- Scarisbrick, I.A.; Asakura, K.; Rodriguez, M. Neurotrophin-4/5 Promotes Proliferation of Oligodendrocyte-Type-2 Astrocytes (O-2A). Dev. Brain Res. 2000, 123, 87–90.
- Kondo, T.; Raff, M.C. A Role for Noggin in the Development of Oligodendrocyte Precursor Cells. Dev. Biol. 2004, 267, 242–251.
- Cohen, R.I.; Marmur, R.; Norton, W.T.; Mehler, M.F.; Kessler, J.A. Nerve Growth Factor and Neurotrophin-3 Differentially Regulate the Proliferation and Survival of Developing Rat Brain Oligodendrocytes. J. Neurosci. 1996, 16, 6433–6442.
- Castro, D.S.; Martynoga, B.; Parras, C.; Ramesh, V.; Pacary, E.; Johnston, C.; Drechsel, D.; Lebel-Potter, M.; Garcia, L.G.; Hunt, C.; et al. A Novel Function of the Proneural Factor Ascl1 in Progenitor Proliferation Identified by Genome-Wide Characterization of Its Targets. Genes Dev. 2011, 25, 930–945.
- Warren, N.; Price, D.J. Roles of Pax-6 in Murine Diencephalic Development. Dev. Camb. Engl. 1997, 124, 1573–1582.
- Veiga, S.; Ly, J.; Chan, P.H.; Bresnahan, J.C.; Beattie, M.S. SOD1 Overexpression Improves Features of the Oligodendrocyte Precursor Response in Vitro. Neurosci. Lett. 2011, 503, 10–14.
- Hu, J.; Deng, L.; Wang, X.; Xu, X.-M. Effects of Extracellular Matrix Molecules on the Growth Properties of Oligodendrocyte Progenitor Cells in Vitro. J. Neurosci. Res. 2009, 87, 2854–2862.
- Blaschuk, K.L.; Frost, E.E.; ffrench-Constant, C. The Regulation of Proliferation and Differentiation in Oligodendrocyte Progenitor Cells by AlphaV Integrins. Dev. Camb. Engl. 2000, 127, 1961–1969.
- Adams, K.L.; Riparini, G.; Banerjee, P.; Breur, M.; Bugiani, M.; Gallo, V. Endothelin-1 Signaling Maintains Glial Progenitor Proliferation in the Postnatal Subventricular Zone. Nat. Commun. 2020, 11, 2138.
- Chapman, H.; Riesenberg, A.; Ehrman, L.A.; Kohli, V.; Nardini, D.; Nakafuku, M.; Campbell, K.; Waclaw, R.R. Gsx Transcription Factors Control Neuronal versus Glial Specification in Ventricular Zone Progenitors of the Mouse Lateral Ganglionic Eminence. Dev. Biol. 2018, 442, 115–126.
- Stancic, M.; Slijepcevic, D.; Nomden, A.; Vos, M.J.; de Jonge, J.C.; Sikkema, A.H.; Gabius, H.-J.; Hoekstra, D.; Baron, W. Galectin-4, a Novel Neuronal Regulator of Myelination. Glia 2012, 60, 919–935.
- Canoll, P.D.; Musacchio, J.M.; Hardy, R.; Reynolds, R.; Marchionni, M.A.; Salzer, J.L. GGF/Neuregulin Is a Neuronal Signal That Promotes the Proliferation and Survival and Inhibits the Differentiation of Oligodendrocyte Progenitors. Neuron 1996, 17, 229–243.
- Zhang, S.; Zhu, X.; Gui, X.; Croteau, C.; Song, L.; Xu, J.; Wang, A.; Bannerman, P.; Guo, F. Sox2 Is Essential for Oligodendroglial Proliferation and Differentiation during Postnatal Brain Myelination and CNS Remyelination. J. Neurosci. 2018, 38, 1802–1820.
- Brinkmann, B.G.; Agarwal, A.; Sereda, M.W.; Garratt, A.N.; Müller, T.; Wende, H.; Stassart, R.M.; Nawaz, S.; Humml, C.; Velanac, V.; et al. Neuregulin-1/ErbB Signaling Serves Distinct Functions in Myelination of the Peripheral and Central Nervous System. Neuron 2008, 59, 581–595.
- Dai, J.; Bercury, K.K.; Ahrendsen, J.T.; Macklin, W.B. Olig1 Function Is Required for Oligodendrocyte Differentiation in the Mouse Brain. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 4386–4402.
- See, J.; Zhang, X.; Eraydin, N.; Mun, S.-B.; Mamontov, P.; Golden, J.A.; Grinspan, J.B. Oligodendrocyte Maturation Is Inhibited by Bone Morphogenetic Protein. Mol. Cell. Neurosci. 2004, 26, 481–492.
- Harnisch, K.; Teuber-Hanselmann, S.; Macha, N.; Mairinger, F.; Fritsche, L.; Soub, D.; Meinl, E.; Junker, A. Myelination in Multiple Sclerosis Lesions Is Associated with Regulation of Bone Morphogenetic Protein 4 and Its Antagonist Noggin. Int. J. Mol. Sci. 2019, 20, 154.
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human Oligodendrocytes Derived from Embryonic Stem Cells: Effect of Noggin on Phenotypic Differentiation in vitro and on Myelination in vivo. Mol. Cell. Neurosci. 2007, 34, 310–323.
- Sugimori, M.; Nagao, M.; Parras, C.M.; Nakatani, H.; Lebel, M.; Guillemot, F.; Nakafuku, M. Ascl1 Is Required for Oligodendrocyte Development in the Spinal Cord. Dev. Camb. Engl. 2008, 135, 1271–1281.
- Matsumoto, S.; Banine, F.; Feistel, K.; Foster, S.; Xing, R.; Struve, J.; Sherman, L.S. Brg1 Directly Regulates Olig2 Transcription and Is Required for Oligodendrocyte Progenitor Cell Specification. Dev. Biol. 2016, 413, 173–187.
- Wedel, M.; Fröb, F.; Elsesser, O.; Wittmann, M.-T.; Lie, D.C.; Reis, A.; Wegner, M. Transcription Factor Tcf4 Is the Preferred Heterodimerization Partner for Olig2 in Oligodendrocytes and Required for Differentiation. Nucleic Acids Res. 2020, 48, 4839–4857.
- Zhao, C.; Deng, Y.; Liu, L.; Yu, K.; Zhang, L.; Wang, H.; He, X.; Wang, J.; Lu, C.; Wu, L.N.; et al. Dual Regulatory Switch through Interactions of Tcf7l2/Tcf4 with Stage-Specific Partners Propels Oligodendroglial Maturation. Nat. Commun. 2016, 7, 10883.
- Zhang, K.; Chen, S.; Yang, Q.; Guo, S.; Chen, Q.; Liu, Z.; Li, L.; Jiang, M.; Li, H.; Hu, J.; et al. The Oligodendrocyte Transcription Factor 2 OLIG2 Regulates Transcriptional Repression during Myelinogenesis in Rodents. Nat. Commun. 2022, 13, 1423.
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The Oligodendrocyte-Specific G Protein–Coupled Receptor GPR17 Is a Cell-Intrinsic Timer of Myelination. Nat. Neurosci. 2009, 12, 1398–1406.
- Marin-Husstege, M.; He, Y.; Li, J.; Kondo, T.; Sablitzky, F.; Casaccia-Bonnefil, P. Multiple Roles of Id4 in Developmental Myelination: Predicted Outcomes and Unexpected Findings. Glia 2006, 54, 285–296.
- Huang, H.; Wu, H.; He, W.; Zhou, F.; Yu, X.; Yi, M.; Du, J.; Xie, B.; Qiu, M. Id2 and Id4 Are Not the Major Negative Regulators of Oligodendrocyte Differentiation during Early Central Nervous System Development. Glia 2022, 70, 590–601.
- Swiss, V.A.; Nguyen, T.; Dugas, J.; Ibrahim, A.; Barres, B.; Androulakis, I.P.; Casaccia, P. Identification of a Gene Regulatory Network Necessary for the Initiation of Oligodendrocyte Differentiation. PLoS ONE 2011, 6, e18088.
- Bujalka, H.; Koenning, M.; Jackson, S.; Perreau, V.M.; Pope, B.; Hay, C.M.; Mitew, S.; Hill, A.F.; Lu, Q.R.; Wegner, M.; et al. MYRF Is a Membrane-Associated Transcription Factor That Autoproteolytically Cleaves to Directly Activate Myelin Genes. PLoS Biol. 2013, 11, e1001625.
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin Gene Regulatory Factor Is a Critical Transcriptional Regulator Required for CNS Myelination. Cell 2009, 138, 172–185.
- Hoffmann, S.A.; Hos, D.; Küspert, M.; Lang, R.A.; Lovell-Badge, R.; Wegner, M.; Reiprich, S. Stem Cell Factor Sox2 and Its Close Relative Sox3 Have Differentiation Functions in Oligodendrocytes. Development 2014, 141, 39–50.
- Stolt, C.C.; Lommes, P.; Friedrich, R.P.; Wegner, M. Transcription Factors Sox8 and Sox10 Perform Non-Equivalent Roles during Oligodendrocyte Development despite Functional Redundancy. Development 2004, 131, 2349–2358.
- Stolt, C.C.; Rehberg, S.; Ader, M.; Lommes, P.; Riethmacher, D.; Schachner, M.; Bartsch, U.; Wegner, M. Terminal Differentiation of Myelin-Forming Oligodendrocytes Depends on the Transcription Factor Sox10. Genes Dev. 2002, 16, 165–170.
- Takada, N.; Kucenas, S.; Appel, B. Sox10 Is Necessary for Oligodendrocyte Survival Following Axon Wrapping. Glia 2010, 58, 996–1006.
- Zhang, C.; Huang, H.; Chen, Z.; Zhang, Z.; Lu, W.; Qiu, M. The Transcription Factor NKX2-2 Regulates Oligodendrocyte Differentiation through Domain-Specific Interactions with Transcriptional Corepressors. J. Biol. Chem. 2020, 295, 1879–1888.
- Qi, Y.; Cai, J.; Wu, Y.; Wu, R.; Lee, J.; Fu, H.; Rao, M.; Sussel, L.; Rubenstein, J.; Qiu, M. Control of Oligodendrocyte Differentiation by the Nkx2.2 Homeodomain Transcription Factor. Dev. Camb. Engl. 2001, 128, 2723–2733.
- Ji, S.; Doucette, J.R.; Nazarali, A.J. Sirt2 Is a Novel in Vivo Downstream Target of Nkx2.2 and Enhances Oligodendroglial Cell Differentiation. J. Mol. Cell Biol. 2011, 3, 351–359.
- Hisahara, S.; Iwahara, N.; Matsushita, T.; Suzuki, S.; Matsumura, A.; Fujikura, M.; Yokokawa, K.; Saito, T.; Manabe, T.; Kawamata, J.; et al. SIRT1 Decelerates Morphological Processing of Oligodendrocyte Cell Lines and Regulates the Expression of Cytoskeleton-Related Oligodendrocyte Proteins. Biochem. Biophys. Res. Commun. 2021, 546, 7–14.
- Laitman, B.M.; Asp, L.; Mariani, J.N.; Zhang, J.; Liu, J.; Sawai, S.; Chapouly, C.; Horng, S.; Kramer, E.G.; Mitiku, N.; et al. The Transcriptional Activator Krüppel-like Factor-6 Is Required for CNS Myelination. PLOS Biol. 2016, 14, e1002467.
- Vondran, M.W.; Clinton-Luke, P.; Honeywell, J.Z.; Dreyfus, C.F. BDNF+/- Mice Exhibit Deficits in Oligodendrocyte Lineage Cells of the Basal Forebrain. Glia 2010, 58, 848–856.
- Heinrich, M.; Gorath, M.; Richter-Landsberg, C. Neurotrophin-3 (NT-3) Modulates Early Differentiation of Oligodendrocytes in Rat Brain Cortical Cultures. Glia 1999, 28, 244–255.
- Yan, H.; Wood, P.M. NT-3 Weakly Stimulates Proliferation of Adult Rat O1(−)O4(+) Oligodendrocyte-Lineage Cells and Increases Oligodendrocyte Myelination in vitro. J. Neurosci. Res. 2000, 62, 329–335.
- McMorris, F.A.; Dubois-Dalcq, M. Insulin-like Growth Factor I Promotes Cell Proliferation and Oligodendroglial Commitment in Rat Glial Progenitor Cells Developing in vitro. J. Neurosci. Res. 1988, 21, 199–209.
- Zhou, L.; Shao, C.-Y.; Xie, Y.-J.; Wang, N.; Xu, S.-M.; Luo, B.-Y.; Wu, Z.-Y.; Ke, Y.H.; Qiu, M.; Shen, Y. Gab1 Mediates PDGF Signaling and Is Essential to Oligodendrocyte Differentiation and CNS Myelination. eLife 2020, 9, e52056.
- Zhou, X.; Shin, S.; He, C.; Zhang, Q.; Rasband, M.N.; Ren, J.; Dai, C.; Zorrilla-Veloz, R.I.; Shingu, T.; Yuan, L.; et al. Qki Regulates Myelinogenesis through Srebp2-Dependent Cholesterol Biosynthesis. eLife 2021, 10, e60467.
- Ho, W.Y.; Chang, J.-C.; Lim, K.; Cazenave-Gassiot, A.; Nguyen, A.T.; Foo, J.C.; Muralidharan, S.; Viera-Ortiz, A.; Ong, S.J.M.; Hor, J.H.; et al. TDP-43 Mediates SREBF2-Regulated Gene Expression Required for Oligodendrocyte Myelination. J. Cell Biol. 2021, 220, e201910213.
- Morita, J.; Kano, K.; Kato, K.; Takita, H.; Sakagami, H.; Yamamoto, Y.; Mihara, E.; Ueda, H.; Sato, T.; Tokuyama, H.; et al. Structure and Biological Function of ENPP6, a Choline-Specific Glycerophosphodiester-Phosphodiesterase. Sci. Rep. 2016, 6, 20995.
- Baas, D.; Bourbeau, D.; Sarliève, L.L.; Ittel, M.E.; Dussault, J.H.; Puymirat, J. Oligodendrocyte Maturation and Progenitor Cell Proliferation Are Independently Regulated by Thyroid Hormone. Glia 1997, 19, 324–332.
- Barres, B.A.; Lazar, M.A.; Raff, M.C. A Novel Role for Thyroid Hormone, Glucocorticoids and Retinoic Acid in Timing Oligodendrocyte Development. Dev. Camb. Engl. 1994, 120, 1097–1108.
- Baas, D.; Fressinaud, C.; Ittel, M.E.; Reeber, A.; Dalençon, D.; Puymirat, J.; Sarliève, L.L. Expression of Thyroid Hormone Receptor Isoforms in Rat Oligodendrocyte Cultures. Effect of 3,5,3′-Triiodo-l-Thyronine. Neurosci. Lett. 1994, 176, 47–51.
- Kondo, T.; Raff, M. Basic Helix-Loop-Helix Proteins and the Timing of Oligodendrocyte Differentiation. Dev. Camb. Engl. 2000, 127, 2989–2998.
- Genoud, S.; Lappe-Siefke, C.; Goebbels, S.; Radtke, F.; Aguet, M.; Scherer, S.S.; Suter, U.; Nave, K.-A.; Mantei, N. Notch1 Control of Oligodendrocyte Differentiation in the Spinal Cord. J. Cell Biol. 2002, 158, 709–718.
- Hu, Q.-D.; Ang, B.-T.; Karsak, M.; Hu, W.-P.; Cui, X.-Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.-K.; et al. F3/Contactin Acts as a Functional Ligand for Notch during Oligodendrocyte Maturation. Cell 2003, 115, 163–175.
- Emery, B. Regulation of Oligodendrocyte Differentiation and Myelination. Science 2010, 330, 779–782.
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. LINGO-1, a Transmembrane Signaling Protein, Inhibits Oligodendrocyte Differentiation and Myelination through Intercellular Self-Interactions. J. Biol. Chem. 2012, 287, 22184–22195.
- Hirahara, Y.; Bansal, R.; Honke, K.; Ikenaka, K.; Wada, Y. Sulfatide Is a Negative Regulator of Oligodendrocyte Differentiation: Development in Sulfatide-Null Mice. Glia 2004, 45, 269–277.
- Choi, M.-H.; Na, J.E.; Yoon, Y.R.; Rhyu, I.J.; Ko, Y.-G.; Baik, J.-H. Hypomyelination and Cognitive Impairment in Mice Lacking CD133 (Prominin-1). Biochem. Biophys. Res. Commun. 2018, 502, 291–298.
- Brown, T.L.; Hashimoto, H.; Finseth, L.T.; Wood, T.L.; Macklin, W.B. PAK1 Positively Regulates Oligodendrocyte Morphology and Myelination. J. Neurosci. 2021, 41, 1864–1877.
- Quarles, R.H. Myelin-Associated Glycoprotein (MAG): Past, Present and Beyond. J. Neurochem. 2007, 100, 1431–1448.
- Stankoff, B.; Aigrot, M.-S.; Noël, F.; Wattilliaux, A.; Zalc, B.; Lubetzki, C. Ciliary Neurotrophic Factor (CNTF) Enhances Myelin Formation: A Novel Role for CNTF and CNTF-Related Molecules. J. Neurosci. 2002, 22, 9221–9227.
- Kalsi, A.S.; Greenwood, K.; Wilkin, G.; Butt, A.M. Kir4.1 Expression by Astrocytes and Oligodendrocytes in CNS White Matter: A Developmental Study in the Rat Optic Nerve. J. Anat. 2004, 204, 475–485.
- Howng, S.Y.B.; Avila, R.L.; Emery, B.; Traka, M.; Lin, W.; Watkins, T.; Cook, S.; Bronson, R.; Davisson, M.; Barres, B.A.; et al. ZFP191 Is Required by Oligodendrocytes for CNS Myelination. Genes Dev. 2010, 24, 301–311.
- Müller, R.; Heinrich, M.; Heck, S.; Blohm, D.; Richter-Landsberg, C. Expression of Microtubule-Associated Proteins MAP2 and Tau in Cultured Rat Brain Oligodendrocytes. Cell Tissue Res. 1997, 288, 239–249.
- Chen, X.; Ku, L.; Mei, R.; Liu, G.; Xu, C.; Wen, Z.; Zhao, X.; Wang, F.; Xiao, L.; Feng, Y. Novel Schizophrenia Risk Factor Pathways Regulate FEZ1 to Advance Oligodendroglia Development. Transl. Psychiatry 2017, 7, 1293.
- Lee, J.; Gravel, M.; Zhang, R.; Thibault, P.; Braun, P.E. Process Outgrowth in Oligodendrocytes Is Mediated by CNP, a Novel Microtubule Assembly Myelin Protein. J. Cell Biol. 2005, 170, 661–673.
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione Deficit Impairs Myelin Maturation: Relevance for White Matter Integrity in Schizophrenia Patients. Mol. Psychiatry 2015, 20, 827–838.
- Lee, X.; Hu, Y.; Zhang, Y.; Yang, Z.; Shao, Z.; Qiu, M.; Pepinsky, B.; Miller, R.H.; Mi, S. Oligodendrocyte Differentiation and Myelination Defects in OMgp Null Mice. Mol. Cell. Neurosci. 2011, 46, 752–761.
- Kaji, S.; Maki, T.; Ueda, J.; Ishimoto, T.; Inoue, Y.; Yasuda, K.; Sawamura, M.; Hikawa, R.; Ayaki, T.; Yamakado, H.; et al. BCAS1-Positive Immature Oligodendrocytes Are Affected by the α-Synuclein-Induced Pathology of Multiple System Atrophy. Acta Neuropathol. Commun. 2020, 8, 120.
- Fard, M.K.; van der Meer, F.; Sánchez, P.; Cantuti-Castelvetri, L.; Mandad, S.; Jäkel, S.; Fornasiero, E.F.; Schmitt, S.; Ehrlich, M.; Starost, L.; et al. BCAS1 Expression Defines a Population of Early Myelinating Oligodendrocytes in Multiple Sclerosis Lesions. Sci. Transl. Med. 2017, 9, eaam7816.
- Ikenaka, K.; Kagawa, T.; Mikoshiba, K. Selective Expression of DM-20, an Alternatively Spliced Myelin Proteolipid Protein Gene Product, in Developing Nervous System and in Nonglial Cells. J. Neurochem. 1992, 58, 2248–2253.
- Galiano, M.R.; Andrieux, A.; Deloulme, J.C.; Bosc, C.; Schweitzer, A.; Job, D.; Hallak, M.E. Myelin Basic Protein Functions as a Microtubule Stabilizing Protein in Differentiated Oligodendrocytes. J. Neurosci. Res. 2006, 84, 534–541.
- Uhlenberg, B.; Schuelke, M.; Rüschendorf, F.; Ruf, N.; Kaindl, A.M.; Henneke, M.; Thiele, H.; Stoltenburg-Didinger, G.; Aksu, F.; Topaloğlu, H.; et al. Mutations in the Gene Encoding Gap Junction Protein Alpha 12 (Connexin 46.6) Cause Pelizaeus-Merzbacher-like Disease. Am. J. Hum. Genet. 2004, 75, 251–260.
- Menichella, D.M.; Goodenough, D.A.; Sirkowski, E.; Scherer, S.S.; Paul, D.L. Connexins Are Critical for Normal Myelination in the CNS. J. Neurosci. 2003, 23, 5963–5973.
- Wasseff, S.K.; Scherer, S.S. Cx32 and Cx47 Mediate Oligodendrocyte:Astrocyte and Oligodendrocyte:Oligodendrocyte Gap Junction Coupling. Neurobiol. Dis. 2011, 42, 506–513.
- Mosser, J.; Douar, A.M.; Sarde, C.O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.M.; Mandel, J.L.; Aubourg, P. Putative X-Linked Adrenoleukodystrophy Gene Shares Unexpected Homology with ABC Transporters. Nature 1993, 361, 726–730.
- Strachan, L.R.; Stevenson, T.J.; Freshner, B.; Keefe, M.D.; Miranda Bowles, D.; Bonkowsky, J.L. A Zebrafish Model of X-Linked Adrenoleukodystrophy Recapitulates Key Disease Features and Demonstrates a Developmental Requirement for Abcd1 in Oligodendrocyte Patterning and Myelination. Hum. Mol. Genet. 2017, 26, 3600–3614.
- Schnädelbach, O.; Ozen, I.; Blaschuk, O.W.; Meyer, R.L.; Fawcett, J.W. N-Cadherin Is Involved in Axon-Oligodendrocyte Contact and Myelination. Mol. Cell. Neurosci. 2001, 17, 1084–1093.
- Laursen, L.S.; Chan, C.W.; ffrench-Constant, C. An Integrin–Contactin Complex Regulates CNS Myelination by Differential Fyn Phosphorylation. J. Neurosci. 2009, 29, 9174–9185.
- Charles, P.; Tait, S.; Faivre-Sarrailh, C.; Barbin, G.; Gunn-Moore, F.; Denisenko-Nehrbass, N.; Guennoc, A.-M.; Girault, J.-A.; Brophy, P.J.; Lubetzki, C. Neurofascin Is a Glial Receptor for the Paranodin/Caspr-Contactin Axonal Complex at the Axoglial Junction. Curr. Biol. 2002, 12, 217–220.
- Thaxton, C.; Pillai, A.M.; Pribisko, A.L.; Labasque, M.; Dupree, J.L.; Faivre-Sarrailh, C.; Bhat, M.A. In Vivo Deletion of Immunoglobulin Domains 5 and 6 in Neurofascin (Nfasc) Reveals Domain-Specific Requirements in Myelinated Axons. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 4868–4876.
- Labasque, M.; Hivert, B.; Nogales-Gadea, G.; Querol, L.; Illa, I.; Faivre-Sarrailh, C. Specific Contactin N-Glycans Are Implicated in Neurofascin Binding and Autoimmune Targeting in Peripheral Neuropathies. J. Biol. Chem. 2014, 289, 7907–7918.
- Klingseisen, A.; Ristoiu, A.-M.; Kegel, L.; Sherman, D.L.; Rubio-Brotons, M.; Almeida, R.G.; Koudelka, S.; Benito-Kwiecinski, S.K.; Poole, R.J.; Brophy, P.J.; et al. Oligodendrocyte Neurofascin Independently Regulates Both Myelin Targeting and Sheath Growth in the CNS. Dev. Cell 2019, 51, 730–744.e6.
- Çolakoğlu, G.; Bergstrom-Tyrberg, U.; Berglund, E.O.; Ranscht, B. Contactin-1 Regulates Myelination and Nodal/Paranodal Domain Organization in the Central Nervous System. Proc. Natl. Acad. Sci. USA 2014, 111, E394–E403.
- Traka, M.; Goutebroze, L.; Denisenko, N.; Bessa, M.; Nifli, A.; Havaki, S.; Iwakura, Y.; Fukamauchi, F.; Watanabe, K.; Soliven, B.; et al. Association of TAG-1 with Caspr2 Is Essential for the Molecular Organization of Juxtaparanodal Regions of Myelinated Fibers. J. Cell Biol. 2003, 162, 1161–1172.
- Elazar, N.; Vainshtein, A.; Golan, N.; Vijayaragavan, B.; Schaeren-Wiemers, N.; Eshed-Eisenbach, Y.; Peles, E. Axoglial Adhesion by Cadm4 Regulates CNS Myelination. Neuron 2019, 101, 224–231.e5.
- Hughes, A.N.; Appel, B. Oligodendrocytes Express Synaptic Proteins That Modulate Myelin Sheath Formation. Nat. Commun. 2019, 10, 4125.
- Sturgill, E.R.; Aoki, K.; Lopez, P.H.; Colacurcio, D.; Vajn, K.; Lorenzini, I.; Majić, S.; Yang, W.H.; Heffer, M.; Tiemeyer, M.; et al. Biosynthesis of the Major Brain Gangliosides GD1a and GT1b. Glycobiology 2012, 22, 1289–1301.
- Schnaar, R.L. Brain Gangliosides in Axon-Myelin Stability and Axon Regeneration. FEBS Lett. 2010, 584, 1741–1747.
- Harboe, M.; Torvund-Jensen, J.; Kjaer-Sorensen, K.; Laursen, L.S. Ephrin-A1-EphA4 Signaling Negatively Regulates Myelination in the Central Nervous System. Glia 2018, 66, 934–950.
- Linneberg, C.; Harboe, M.; Laursen, L.S. Axo-Glia Interaction Preceding CNS Myelination Is Regulated by Bidirectional Eph-Ephrin Signaling. ASN Neuro 2015, 7, 175909141560285.
- Almeida, R.G. The Rules of Attraction in Central Nervous System Myelination. Front. Cell. Neurosci. 2018, 12, 367.
- Charles, P.; Hernandez, M.P.; Stankoff, B.; Aigrot, M.S.; Colin, C.; Rougon, G.; Zalc, B.; Lubetzki, C. Negative Regulation of Central Nervous System Myelination by Polysialylated-Neural Cell Adhesion Molecule. Proc. Natl. Acad. Sci. USA 2000, 97, 7585–7590.
- Redmond, S.A.; Mei, F.; Eshed-Eisenbach, Y.; Osso, L.A.; Leshkowitz, D.; Shen, Y.-A.A.; Kay, J.N.; Aurrand-Lions, M.; Lyons, D.A.; Peles, E.; et al. Somatodendritic Expression of JAM2 Inhibits Oligodendrocyte Myelination. Neuron 2016, 91, 824–836.
- Díez-Revuelta, N.; Higuero, A.M.; Velasco, S.; Peñas-de-la-Iglesia, M.; Gabius, H.-J.; Abad-Rodríguez, J. Neurons Define Non-Myelinated Axon Segments by the Regulation of Galectin-4-Containing Axon Membrane Domains. Sci. Rep. 2017, 7, 12246.
- Xin, M.; Yue, T.; Ma, Z.; Wu, F.; Gow, A.; Lu, Q.R. Myelinogenesis and Axonal Recognition by Oligodendrocytes in Brain Are Uncoupled in Olig1-Null Mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1354–1365.
- Lüders, K.A.; Nessler, S.; Kusch, K.; Patzig, J.; Jung, R.B.; Möbius, W.; Nave, K.-A.; Werner, H.B. Maintenance of High Proteolipid Protein Level in Adult Central Nervous System Myelin Is Required to Preserve the Integrity of Myelin and Axons. Glia 2019, 67, 634–649.
- Chow, E.; Mottahedeh, J.; Prins, M.; Ridder, W.; Nusinowitz, S.; Bronstein, J.M. Disrupted Compaction of CNS Myelin in an OSP/Claudin-11 and PLP/DM20 Double Knockout Mouse. Mol. Cell. Neurosci. 2005, 29, 405–413.
- Schachner, M.; Bartsch, U. Multiple Functions of the Myelin-Associated Glycoprotein MAG (Siglec-4a) in Formation and Maintenance of Myelin. Glia 2000, 29, 154–165.
- Clements, C.S.; Reid, H.H.; Beddoe, T.; Tynan, F.E.; Perugini, M.A.; Johns, T.G.; Bernard, C.C.A.; Rossjohn, J. The Crystal Structure of Myelin Oligodendrocyte Glycoprotein, a Key Autoantigen in Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 11059–11064.
- Johns, T.G.; Bernard, C.C.A. The Structure and Function of Myelin Oligodendrocyte Glycoprotein. J. Neurochem. 1999, 72, 1–9.
- Rasband, M.N.; Tayler, J.; Kaga, Y.; Yang, Y.; Lappe-Siefke, C.; Nave, K.-A.; Bansal, R. CNP Is Required for Maintenance of Axon-Glia Interactions at Nodes of Ranvier in the CNS. Glia 2005, 50, 86–90.
- Schäfer, I.; Müller, C.; Luhmann, H.J.; White, R. MOBP Levels Are Regulated by Fyn Kinase and Affect the Morphological Differentiation of Oligodendrocytes. J. Cell Sci. 2016, 129, 930–942.
- Vourc’h, P.; Andres, C. Oligodendrocyte Myelin Glycoprotein (OMgp): Evolution, Structure and Function. Brain Res. Rev. 2004, 45, 115–124.
- Chang, K.-J.; Susuki, K.; Dours-Zimmermann, M.T.; Zimmermann, D.R.; Rasband, M.N. Oligodendrocyte Myelin Glycoprotein Does Not Influence Node of Ranvier Structure or Assembly. J. Neurosci. 2010, 30, 14476–14481.
- Koenning, M.; Jackson, S.; Hay, C.M.; Faux, C.; Kilpatrick, T.J.; Willingham, M.; Emery, B. Myelin Gene Regulatory Factor Is Required for Maintenance of Myelin and Mature Oligodendrocyte Identity in the Adult CNS. J. Neurosci. 2012, 32, 12528–12542.
- Cai, J.; Zhu, Q.; Zheng, K.; Li, H.; Qi, Y.; Cao, Q.; Qiu, M. Co-Localization of Nkx6.2 and Nkx2.2 Homeodomain Proteins in Differentiated Myelinating Oligodendrocytes. Glia 2010, 58, 458–468.
- Huang, H.; Teng, P.; Du, J.; Meng, J.; Hu, X.; Tang, T.; Zhang, Z.; Qi, Y.B.; Qiu, M. Interactive Repression of MYRF Self-Cleavage and Activity in Oligodendrocyte Differentiation by TMEM98 Protein. J. Neurosci. 2018, 38, 9829–9839.
- Othman, A.; Frim, D.M.; Polak, P.; Vujicic, S.; Arnason, B.G.W.; Boullerne, A.I. Olig1 Is Expressed in Human Oligodendrocytes during Maturation and Regeneration. Glia 2011, 59, 914–926.
- Ishii, A.; Furusho, M.; Dupree, J.L.; Bansal, R. Role of ERK1/2 MAPK Signaling in the Maintenance of Myelin and Axonal Integrity in the Adult CNS. J. Neurosci. 2014, 34, 16031–16045.
- Furusho, M.; Ishii, A.; Bansal, R. Signaling by FGF Receptor 2, Not FGF Receptor 1, Regulates Myelin Thickness through Activation of ERK1/2–MAPK, Which Promotes MTORC1 Activity in an Akt-Independent Manner. J. Neurosci. 2017, 37, 2931–2946.
- Zhang, L.; He, X.; Liu, L.; Jiang, M.; Zhao, C.; Wang, H.; He, D.; Zheng, T.; Zhou, X.; Hassan, A.; et al. Hdac3 Interaction with P300 Histone Acetyltransferase Regulates the Oligodendrocyte and Astrocyte Lineage Fate Switch. Dev. Cell 2016, 36, 316–330.
- Marcus, J.; Honigbaum, S.; Shroff, S.; Honke, K.; Rosenbluth, J.; Dupree, J.L. Sulfatide Is Essential for the Maintenance of CNS Myelin and Axon Structure. Glia 2006, 53, 372–381.
- Coetzee, T.; Dupree, J.L.; Popko, B. Demyelination and Altered Expression of Myelin-Associated Glycoprotein Isoforms in the Central Nervous System of Galactolipid-Deficient Mice. J. Neurosci. Res. 1998, 54, 613–622.
- Hulshagen, L.; Krysko, O.; Bottelbergs, A.; Huyghe, S.; Klein, R.; Van Veldhoven, P.P.; De Deyn, P.P.; D’Hooge, R.; Hartmann, D.; Baes, M. Absence of Functional Peroxisomes from Mouse CNS Causes Dysmyelination and Axon Degeneration. J. Neurosci. 2008, 28, 4015–4027.
- Morita, M.; Shinbo, S.; Asahi, A.; Imanaka, T. Very Long Chain Fatty Acid β-Oxidation in Astrocytes: Contribution of the ABCD1-Dependent and -Independent Pathways. Biol. Pharm. Bull. 2012, 35, 1972–1979.
- Sato, Y.; Yoshikawa, F.; Sadakata, T.; Shinoda, Y.; Koebis, M.; Furuichi, T. Age-Dependent Redistribution and Hypersialylation of the Central Myelin Paranodal Loop Membrane Protein Opalin in the Mouse Brain. Neurosci. Lett. 2014, 581, 14–19.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
04 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No