Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jung Weon Weon Lee | -- | 2607 | 2022-08-02 10:44:47 | | | |
| 2 | Sirius Huang | Meta information modification | 2607 | 2022-08-03 03:14:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kim, J.E.; Kim, E.; Lee, J.W. Roles of TM4SF5 in Hepatocyte Metabolism. Encyclopedia. Available online: https://encyclopedia.pub/entry/25763 (accessed on 24 June 2026).
Kim JE, Kim E, Lee JW. Roles of TM4SF5 in Hepatocyte Metabolism. Encyclopedia. Available at: https://encyclopedia.pub/entry/25763. Accessed June 24, 2026.
Kim, Ji Eon, Eunmi Kim, Jung Weon Lee. "Roles of TM4SF5 in Hepatocyte Metabolism" Encyclopedia, https://encyclopedia.pub/entry/25763 (accessed June 24, 2026).
Kim, J.E., Kim, E., & Lee, J.W. (2022, August 02). Roles of TM4SF5 in Hepatocyte Metabolism. In Encyclopedia. https://encyclopedia.pub/entry/25763
Kim, Ji Eon, et al. "Roles of TM4SF5 in Hepatocyte Metabolism." Encyclopedia. Web. 02 August, 2022.
Copy Citation
Transmembrane 4 L six family member 5 (TM4SF5) has been observed to play roles in the regulation of metabolic functions and activities in hepatocytes using in vitro cell and in vivo animal models without or with TM4SF5 expression in addition to clinical liver tissue samples. TM4SF5 is present on the membranes of different organelles or vesicles and cooperates with transporters for fatty acids, amino acids, and monocarbohydrates, thus regulating nutrient uptake into hepatocytes and metabolism and leading to phenotypes of chronic liver diseases.
hepatocyte
metabolism
protein–protein interaction
nonalcoholic fatty liver disease
steatohepatitis
tetraspan(in)
TM4SF5
1. The TM4SF5-Enriched Microdomain (T5ERM)
Similar to genuine tetraspanin family members, TM4SF5 has four transmembrane (TM) domains: two short cytosolic N- and C-terminal tails and two extracellular loops (SEL and LEL) [1][2]. Meanwhile, the difference between TM4SF5 and the tetraspanins is that TM4SF5 includes no CCG residues and has relatively nonconserved sequences in the LEL. Because other tetraspanins can translocate to cellular membranes, including exosomal membranes, TM4SF5 can also be involved in cellular or environmental processes [3][4][5][6][7]. Tetraspanins form complexes with other receptors at tetraspanin-enriched microdomains (TERMs) or tetraspanin webs for the regulation of diverse cellular functions [8]. Similarly, TM4SF5 also interacts with proteins, including the growth factor receptors EGFR [9], IGFR [10], cytokine receptor (IL-6Rα) [11][12], and integrins [13][14] and also with solute carrier (SLC) family transporters [15][16][17]. The interaction or association of TM4SF5 with diverse membrane proteins (when forming TM4SF5-enriched microdomains, T5ERMs) may, therefore, be dynamically modulated, depending on the intracellular signaling and/or extracellular environment of specific cells. Following dynamic changes in intracellular and/or extracellular metabolic situations, TM4SF5 may change its intracellular localization and binding partners to regulate the expression, stability, binding, and activity of binding partner proteins in T5ERMs in a spatiotemporal manner [18] (Figure 1).
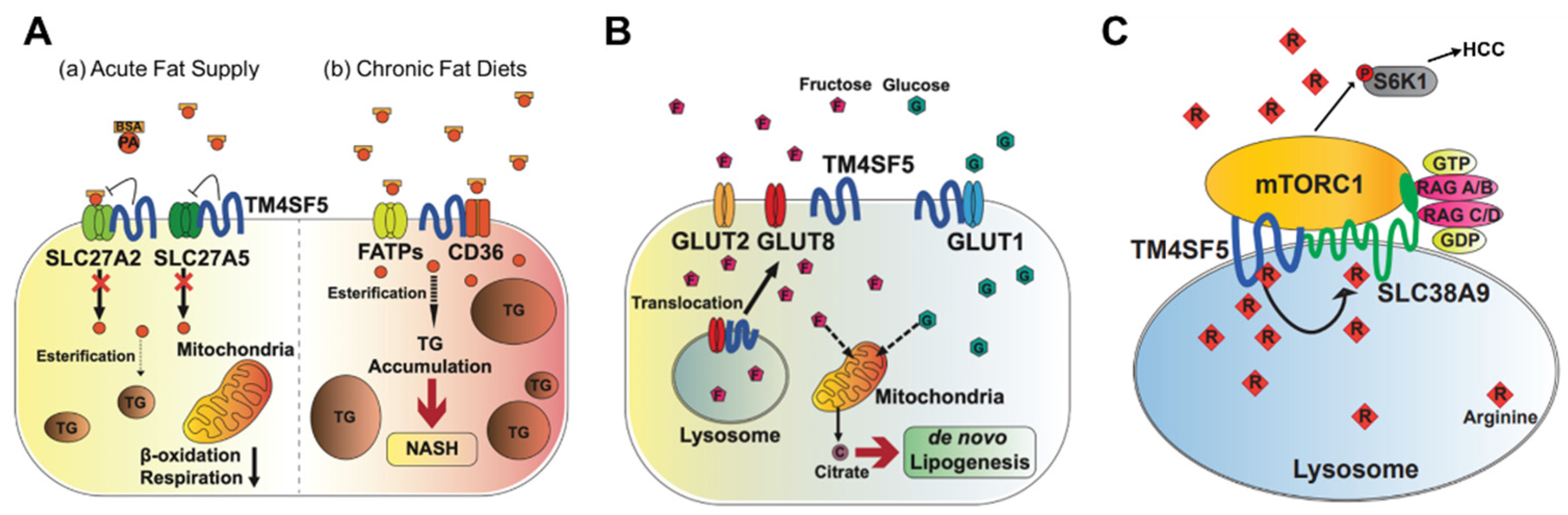
Figure 1. Metabolic regulation by TM4SF5. TM4SF5 can alter the metabolism of lipids, glucose/fructose, and arginine, resulting in the development of various features of chronic liver diseases. (A) Depending on the acute or chronic fatty acid or the fat supply, TM4SF5 in hepatocytes may negatively or positively support the transport activities of the fatty acid transporters via associations with the transporters, respectively. In case of acute supply, hepatocyyte TM4SF5 expression appears to reduce the fatty acid trasporting activity of SLC27A2 and SLC27A5, leading to less fat accumulation in the cells, whereas chronic intake of excess fatty acids leads to TM4SF5-mediated nonalcholoic steatohepatitis due to increased fatty acid uptake and accumulation. (B) When excessive fructose is available, TM4SF5 in hepatocytes causes GLUT8 from endosomal membranes to move toward plasma membranes following its release from their interaction, leading to increased GLUT8-mediated fructose uptake and, eventually, de novo lipogenesis (DNL). (C) Depending on the availability of extracellular (and thereby lysosomal) L-arginine, TM4SF5 binds to and causes mTOR to be translocated to lysosomal membranes, where TM4SF5 localizes and also binds to the L-arginine transporter (SLC38A9, solute carrier family 38 member 9) at the lysosome. The binding affinity of L-arginine to TM4SF5 is greater than that to SLC38A9, leading to the sensing of physiological L-arginine levels in the lysosomal lumen, enough for the export of L-arginine via SLC38A9 to the cytosol. This causes mTOR and S6K1 activation and, eventually, HCC development. FATPs, fatty acid transport proteins; CD36, fatty acid translocase; SREBP1, sterol regulatory element-binding protein 1; GLUT, glucose transporter; mTORC1, mechanistic target of rapamycin complex 1.
2. Regulation of Lipid Metabolism by TM4SF5
Metabolic disturbances cause chronic liver diseases, including the metabolic syndrome NAFLD [19], liver cancer [19], and diabetes [20]. Importantly, the occurrence of liver diseases is associated with the dysregulation of hepatic lipid metabolism [21]. Moreover, chronic liver disease can alter hepatic lipid metabolism, promoting the dysregulation of circulating lipid levels, which contributes to the occurrence of dyslipidemia [22]. In addition, TM4SF5 and interacting proteins in T5ERMs regulate the metabolic status of lipids in hepatocytes and immune cells by affecting multiple levels of lipid metabolism [18]. Possibly via post-translational modifications of N-glycosylation or palmitoylation of TM4SF5 at residues N138 and N155 or nine cysteines near to the transmembrane domians, TM4SF5 binders can influence expression, activity, stability, binding, and/or subcellular localization, eventually leading to modulated activity of the signaling molecules and pathways [9][23]. It is likely that associations of TM4SF5 with other membrane proteins via the transmembrane domains and cytosolic loop and tails with cytosolic proteins are possible [24]. Therefore, at T5ERMs in hepatocytes under certain metabolic conditions, TM4SF5 can have specific binding partners while responding to metabolic needs or the environment.
Triacylglycerols (TGs) are the main constituents of body fat in humans [25]. The TG content in hepatocytes is regulated by the hepatic fatty acid uptake, fatty acid synthesis, esterification, and fatty acid oxidation [26]. Moreover, fatty acids regulate the overall lipid metabolism by binding to nuclear receptors that modulate gene transcription [25]. Nonesterified fatty acids enter cells via transporters (fatty acid transport proteins, FATPs), fatty acid translocases, or diffusion [27]. TM4SF5 interacts with SLC27A2 and SLC27A5, fatty acid transporter members expressed in hepatocytes, to regulate fatty acid uptake and accumulation following an acute supply of fatty acids [15]. Acyl-CoA can be oxidized in either the mitochondria or peroxisomes [28]. The entry of long-chain fatty acids into the mitochondria is regulated by the enzyme CPT1 [29]. The intramitochondrial oxidation of fatty acyl-CoA occurs through β-oxidation pathways, resulting in the formation of acetyl-CoA, which can be completely oxidized to carbon dioxide during the tricarboxylic acid cycle. TM4SF5-positive hepatocytes show decreased β-oxidation activity compared to TM4SF5-negative hepatocytes in vitro following an acute supply of fatty acids [15] (Figure 1A). Although the associations of TM4SF5 with SLC27A members have not been tested under conditions of a chronic fat supply or high-fat diet, it is likely that the acute fatty acid supply situation differs from chronic fatty acid availability in terms of the activity of the fatty acid transporters. Indeed, TM4SF5 knockout-mice fed HFD (60% kCal) for 10 weeks showed increased body weights compared with those of WT mice [30] (Figure 1A).
Fatty acids regulate gene expression by controlling the activity or abundance of key transcription factors [31]. Many transcription factors have been identified as possible targets for fatty acid regulation, including the peroxisome proliferator-activated receptors (alpha, beta, and gamma) SREBP-1c, retinoid X receptor, and LXRα [32]. They integrate signals from various pathways and coordinate the activity of the metabolic machinery necessary for fatty acid metabolism, which is dependent on the supply of energy and fatty acids. TM4SF5-positive hepatocytes exhibit lipid accumulation, decreased Sirtuin1, increased SREBPs, and inactive STAT3, leading to steatotic phenotypes in vitro and in vivo [30]. This relationship appears to become oppositive due to the progression to NASH associated with fibrosis (e.g., TM4SF5 expression leads to increased Sirtuin 1 and active STAT3) [30].
3. Regulation of De Novo Lipogenesis (DNL) by TM4SF5
Hepatic fat accumulation is induced by dietary fat and metabolic conversion of other nutrients. Most importantly, an excessive carbohydrate intake contributes to hepatic lipid accumulation by DNL [33]. The lipogenesis pathway includes a coordinated series of enzymatic reactions [34]. The first step is the conversion of citrate to acetyl-CoA by ATP-citrate lyase (ACLY). The resulting acetyl-CoA is carboxylated to malonyl-CoA by acetyl-CoA carboxylase. The synthesis of palmitate from malonyl-CoA is the key rate-limiting step catalyzed by fatty acid synthase (FASN). The main product of DNL is palmitate, which is further converted into complex fatty acids.
Abnormally elevated DNL is associated with the development of diverse diseases, including metabolic syndrome, type 2 diabetes, hepatic steatosis, and NAFLD [35]. Hepatic DNL is increased in patients with NAFLD, while the contribution of dietary fat and plasma FFA to hepatic lipids does not change significantly [36]. Moreover, the expression levels of ACLY, ACC, and FASN increase in patients with NAFLD [37]. Abnormally increased glucose levels provide substrates for DNL, leading to NAFLD [38]. TM4SF5 interacts with GLUT1, one of the glucose transporters, to facilitate a glucose influx in hepatocytes and macrophages [11]. During excessive fructose consumption, TM4SF5 knockout-mice show reduced fat accumulation in the liver compared with WT mice, indicating that TM4SF5 may facilitate fat accumulation via lipogenesis following fructose uptake via GLUT8 [39] (Figure 1B).
Fructose and glucose induce hepatic DNL [40]. When a high-fructose diet is consumed, fructose is transported by plasma membrane transporters (GLUT2 and GLUT8) in hepatocytes, and fructolysis is initiated by the phosphorylation of fructose by ketohexokinase (KHK) [41]. The expression of TM4SF5 induces hepatic fructose influx by regulating the intracellular localization and fructose-transporting activity of GLUT8 [39]. Molecular studies have shown that TM4SF5 regulates the intracellular localization of GLUT8, which is dependent on fructose treatment, resulting in fructose being transported to intracellular regions and metabolized to fatty acids and lipids. When TM4SF5 releases GLUT8 proximally to the plasma membranes following fructose treatment of the hepatocyte culture media, GLUT8 becomes active for the uptake of fructose but not glucose. The binding of TM4SF5 to GLUT8 is obvious at the endosomal membranes when the culture media lacks fructose. The binding of TM4SF5 to GLUT8 appears to involve V156 and T157 residues very near to the 4th transmembrane domain during coimmunoprecipitation [39].
Tm4sf5 knockout mice show less steatotic phenotypic properties, including less fat accumulation, decreased insulin sensitivity, decreased lipogenic gene expression (ACC, FASN, and SREBP1), and lower KHK expression. Furthermore, a high-fat, high-fructose diet accelerates NAFLD progression. Under conditions of a high-fat diet with excessive fructose consumption (30% w/v fructose with 60% kcal fat diet), the knockout or suppression of TM4SF5 relieves DNL, leading to NAFLD progression in vivo [39]. However, further studies are needed to investigate how TM4SF5 expression leads to the regulation of expression of lipogenic enzymes depending on the hepatocyte metabolic status.
4. Regulation of Arginine Metabolism by TM4SF5
Mechanistic target of rapamycin complex 1 (mTORC1), a central component of nutrient sensing, controls metabolism and growth by activating anabolic processes and inhibiting catabolic processes in response to physiological fluctuations of nutrients [42][43]. mTORC1 activation contributes to the regulation of DNL by increasing SREBP1 transcription [44]. The dysregulation of mTOR signaling can result in many human diseases, including obesity, diabetes, fatty liver diseases, and various types of cancers [45][46].
The signaling pathway involving mTORC1 is activated by amino acids (especially leucine, arginine, and glutamine), insulin, and growth factors. The activation of mTORC1 subsequently activates S6K1 and 4EBP1, which stimulate protein translation and cell growth [47]. The lysosomal arginine transporter, SLC38A9, regulates the exit of amino acids from the lysosomal lumen, but its affinity for arginine inside the lysosomal lumen at 100–200 μM is too low to sense the physiological arginine levels [48][49]. TM4SF5 forms a complex with mTOR and SLC38A9 on lysosomal membranes, which is dependent on the level of arginine [17]. In an arginine-depleted situation, TM4SF5 localizes mostly to the plasma membrane, but upon arginine repletion, the translocation of TM4SF5 to lysosomes occurs, where it binds to mTOR and SLC38A9. In addition, TM4SF5 also binds to arginine and recruits arginine from the luminal pool, thus enabling efflux to the cytosol through SLC38A9 action and eventually through the activation of mTOR/S6K. Instead of SLC38A9, which has a lower binding affinity for lysosomal arginine, TM4SF5 can sense and bind sufficient arginine at physiological levels, leading to the transfer of SLC38A9 for export to metabolic intermediates for biomolecule catabolism and S6K1 activation for translational activation [17] (Figure 1C).
mTOR signaling is enhanced in various types of cancer [50]. Furthermore, TM4SF5 is highly expressed in various cancer types, including liver cancer [51][52]. A higher expression of TM4SF5 alone or in combination with mTOR can be associated with poor recurrence-free survival in patients with liver cancer [17]. The arginine level is critical for cell survival, because it is a precursor of many building blocks of cellular components [53]. Many types of cancer cells, especially HCC, are deficient in argininosuccinate synthase 1 (ASS1), which is a rate-liming enzyme for arginine regeneration [54][55]. ASS1-deficient HCC cells, therefore, depend on external arginine inflow or internal arginine generation through protein degradation in lysosomes. TM4SF5, as a physiological sensor of lysosomal arginine in liver cancer cells, contributes to the SLC38A9-dependent efflux of arginine, so anti-TM4SF5 reagents could be used as a strategy for impairing arginine auxotrophs in HCC [52][56][57].
5. Other Hepatocyte Tetraspanins with Nutrient Transporters
TM4SF5 has a similar membrane topology and conserved features, giving it significant protein–protein interaction and subcellular translocalization capacities, compared with the tetraspanins [1][2]. TM4SF5 and certain members of the tetraspanin family including more than 30 members play regulatory roles in cellular metabolism [58]. Tetraspanins are involved in various biological processes, mainly via interacting with different partner molecules and changing their intracellular localization. Nutrient transporters are ubiquitous components of plasma membranes in all cell types. They are linked to the cells’ needs and can be influenced by environmental factors, such as nutrient starvation and repleshment, to acutely remodel the transporters to promote efficient homeostatic metabolism [59]. Mammalian tetraspanins are well known to be involved in the initiation and progression of various cancer types, including HCC [60]. In particular, certain tetraspanins have been shown to be involved in HCC development, such as TM4SF5 [61]. Certain tetraspanins known as HCC risk factors have been shown to play roles in HCC growth, survival, neoangiogenesis, invasiveness, and migration [60]; they include CD151, TSPAN5, CD9, CD82, CD63, etc. In terms of HCC, abnormal metabolic pathways and activties can be involved in the development of liver maligancies. Since the tetraspanins and TM4SF5 also form protein–protein complexes and are involved in the regulation of signaling activity and stability and intracellular localization of the binders, therapeutic targeting of tetraspanin expression and binding availity may be promising strategies [62]. That is, the roles of the tetraspanins may be targeted to block the development of metabolic fatty liver disease and HCC.
Tetraspanins regulate the metabolic plasticity of cells by associating with nutrient transporters, including GLUT (for glucose), CD36 (for fatty acids), and ASCT2 (for glutamine) [58]. Nutrient transporter-altered localization in cells is dependent on nutrient availability [6][63]. Under conditions of nutrient sufficiency, transporters endocytose, but under conditions of nutrient deficiency, their recycling to the plasma membrane is induced to increase their uptake [64]. CD82 and CD9 have been reported to induce the internalization of the EGFR, which activates mTORC1 signaling [65][66]. In addition, CD9 binds to CD36 and contributes to the oxidization of LDL and its uptake into macrophages [67].
Tetraspanins also alter cellular metabolism via small extracellular vesicles (sEVs). Some small extracellular vesicles are derived from the endosomal network and multi-vesicular bodies [68]. CD9, CD81, CD63, CD151, and TSPAN8 have been found on sEVs and have been widely used as sEV markers [69]. Since the tetraspanins have the capacity to interact homophilically and heterophillically with other membrane proteins and receptors, the packing of tetraspanins into sEVs may play a role in the selective packing of proteins into sEVs and the trophic targeting and fusion of sEVs to the target cells. The association of sEVs with the pathobiology of NAFLD indicates that sEVs are inflammatory drivers of NAFLD, and loading with key modulators, including CD9, CD63, and CD81, occurs in the setting of immune-mediated inflammation [70]. In addition, extracellular vesicles released from hepatocytes upon lipotoxic insult can carry cargo containing lipids, proteins, miRNA, and mitochondrial DNA and can act on target cells to cause inflammatory responses through the activation of macrophages and monocytes [70]. These features could be responsible for organ crosstalk under homeostatic or pathological conditions, leading to chronic liver disease. Being similar to CD9, CD63, and CD81, which are known to be present on the sEVs, it is likely that TM4SF5 is loaded into sEVs released from the liver to play regulatory roles in homeostatic and/or pathological pathways associated with various nutrient metabolism functions.
References
- Wright, M.D.; Ni, J.; Rudy, G.B. The L6 membrane proteins—A new four-transmembrane superfamily. Protein Sci. 2000, 9, 1594–1600.
- Lee, J.W. Transmembrane 4 L Six Family Member 5 (TM4SF5)-Mediated Epithelial-Mesenchymal Transition in Liver Diseases. Int. Rev. Cell Mol. Biol. 2015, 319, 141–163.
- Berditchevski, F. Complexes of tetraspanins with integrins: More than meets the eye. J. Cell Sci. 2001, 114, 4143–4151.
- Charrin, S.; Manie, S.; Billard, M.; Ashman, L.; Gerlier, D.; Boucheix, C.; Rubinstein, E. Multiple levels of interactions within the tetraspanin web. Biochem. Biophys. Res. Commun. 2003, 304, 107–112.
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811.
- Berditchevski, F.; Odintsova, E. Tetraspanins as regulators of protein trafficking. Traffic 2007, 8, 89–96.
- Detchokul, S.; Williams, E.D.; Parker, M.W.; Frauman, A.G. Tetraspanins as regulators of the tumour microenvironment: Implications for metastasis and therapeutic strategies. Br. J. Pharmacol. 2014, 171, 5462–5490.
- Yanez-Mo, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdes, M.; Sanchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446.
- Kim, H.J.; Kwon, S.; Nam, S.H.; Jung, J.W.; Kang, M.; Ryu, J.; Kim, J.E.; Cheong, J.G.; Cho, C.Y.; Kim, S.; et al. Dynamic and coordinated single-molecular interactions at TM4SF5-enriched microdomains guide invasive behaviors in 2- and 3-dimensional environments. FASEB J. 2017, 31, 1461–1481.
- Choi, J.; Kang, M.; Nam, S.H.; Lee, G.H.; Kim, H.J.; Ryu, J.; Cheong, J.G.; Jung, J.W.; Kim, T.Y.; Lee, H.Y.; et al. Bidirectional signaling between TM4SF5 and IGF1R promotes resistance to EGFR kinase inhibitors. Lung Cancer 2015, 90, 22–31.
- Kim, E.; Um, H.; Park, J.; Jung, J.W.; Kim, J.E.; Lee, H.; Shin, E.A.; Pinanga, Y.; Lee, H.; Nam, S.H.; et al. TM4SF5-dependent crosstalk between hepatocytes and macrophages to reprogram the inflammatory environment. Cell Rep. 2021, 37, 110018.
- Ryu, J.; Kang, M.; Lee, M.-S.; Kim, H.-J.; Nam, S.H.; Song, H.E.; Lee, D.; Lee, J.W. Cross Talk between the TM4SF5/Focal Adhesion Kinase and the Interleukin-6/STAT3 Pathways Promotes Immune Escape of Human Liver Cancer Cells. Mol. Cell Biol. 2014, 34, 2946–2960.
- Choi, S.; Lee, S.-A.; Kwak, T.K.; Kim, H.J.; Lee, M.J.; Ye, S.-K.; Kim, S.-H.; Kim, S.; Lee, J.W. Cooperation between integrin α5 and tetraspan TM4SF5 regulates VEGF-mediated angiogenic activity. Blood 2009, 113, 1845–1855.
- Lee, S.Y.; Kim, Y.T.; Lee, M.S.; Kim, Y.B.; Chung, E.; Kim, S.; Lee, J.W. Focal adhesion and actin organization by a cross-talk of TM4SF5 with integrin alpha2 are regulated by serum treatment. Exp. Cell Res. 2006, 312, 2983–2999.
- Park, D.; Kim, E.; Lee, H.; Shin, E.A.; Lee, H.; Lee, J.W. Tetraspanin TM4SF5 in hepatocytes negatively modulates SLC27A transporters during acute fatty acid supply. Arch. Biochem. Biophys. 2021, 710, 109004.
- Kim, J.E.; Kim, H.J.; Jung, J.W.; Song, D.G.; Park, D.; Lee, H.; Um, H.; Park, J.; Nam, S.H.; Cho, M.; et al. TM4SF5-mediated CD44v8-10 splicing variant promotes survival of type II alveolar epithelial cells during idiopathic pulmonary fibrosis. Cell Death Dis. 2019, 10, 645.
- Jung, J.W.; Macalino, S.J.Y.; Cui, M.; Kim, J.E.; Kim, H.J.; Song, D.G.; Nam, S.H.; Kim, S.; Choi, S.; Lee, J.W. Transmembrane 4 L Six Family Member 5 Senses Arginine for mTORC1 Signaling. Cell Metab. 2019, 29, 1306–1319.
- Jung, J.W.; Kim, J.E.; Kim, E.; Lee, J.W. Amino acid transporters as tetraspanin TM4SF5 binding partners. Exp. Mol. Med. 2020, 52, 7–14.
- Watanabe, S.; Yaginuma, R.; Ikejima, K.; Miyazaki, A. Liver diseases and metabolic syndrome. J. Gastroenterol. 2008, 43, 509–518.
- Yu, M.W.; Lin, C.L.; Liu, C.J.; Yang, S.H.; Tseng, Y.L.; Wu, C.F. Influence of Metabolic Risk Factors on Risk of Hepatocellular Carcinoma and Liver-Related Death in Men with Chronic Hepatitis B: A Large Cohort Study. Gastroenterology 2017, 153, 1006–1017.
- Paul, B.; Lewinska, M.; Andersen, J.B. Lipid alterations in chronic liver disease and liver cancer. JHEP. Rep. 2022, 4, 100479.
- Arvind, A.; Osganian, S.A.; Cohen, D.E.; Corey, K.E. Lipid and Lipoprotein Metabolism in Liver Disease. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000.
- Kim, H.J.; Kim, E.; Lee, H.; Jung, J.W.; Kim, J.E.; Pack, C.G.; Lee, J.W. SLAC2B-dependent microtubule acetylation regulates extracellular matrix-mediated intracellular TM4SF5 traffic to the plasma membranes. FASEB J. 2021, 35, e21369.
- Jung, O.; Choi, S.; Jang, S.B.; Lee, S.A.; Lim, S.T.; Choi, Y.J.; Kim, H.J.; Kim, D.H.; Kwak, T.K.; Kim, H.; et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J. Cell Sci. 2012, 125, 5960–5973.
- Bhatt-Wessel, B.; Jordan, T.W.; Miller, J.H.; Peng, L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018, 655, 1–11.
- Ensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Biophys. Acta 2012, 1821, 747–753.
- Black, P.N.; Sandoval, A.; Arias-Barrau, E.; DiRusso, C.C. Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism. Immunol. Endocr. Metab. Agents Med. Chem. 2009, 9, 11–17.
- Li, C.; Li, L.; Lian, J.; Watts, R.; Nelson, R.; Goodwin, B.; Lehner, R. Roles of Acyl-CoA:Diacylglycerol Acyltransferases 1 and 2 in Triacylglycerol Synthesis and Secretion in Primary Hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1080–1091.
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046.
- Ryu, J.; Kim, E.; Kang, M.-K.; Song, D.-G.; Shin, E.-A.; Lee, H.; Jung, J.W.; Nam, S.H.; Kim, J.E.; Kim, H.-J.; et al. Differential TM4SF5-mediated SIRT1 modulation and metabolic signaling in nonalcoholic steatohepatitis progression. J. Pathol. 2021, 253, 55–67.
- Jump, D.B.; Tripathy, S.; Depner, C.M. Fatty acid-regulated transcription factors in the liver. Annu. Rev. Nutr. 2013, 33, 249–269.
- Yoshikawa, T.; Ide, T.; Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Matsuzaka, T.; Yatoh, S.; Kitamine, T.; Okazaki, H.; Tamura, Y.; et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol. Endocrinol. 2003, 17, 1240–1254.
- Sanders, F.W.B.; Acharjee, A.; Walker, C.; Marney, L.; Roberts, L.D.; Imamura, F.; Jenkins, B.; Case, J.; Ray, S.; Virtue, S.; et al. Hepatic steatosis risk is partly driven by increased de novo lipogenesis following carbohydrate consumption. Genome Biol. 2018, 19, 79.
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902.
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468.
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293.
- Gong, Z.; Tas, E.; Yakar, S.; Muzumdar, R. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol. Cell. Endocrinol. 2017, 455, 115–130.
- Chen, L.; Chen, X.W.; Huang, X.; Song, B.L.; Wang, Y.; Wang, Y. Regulation of glucose and lipid metabolism in health and disease. Sci. China Life Sci. 2019, 62, 1420–1458.
- Lee, H.; Kim, E.; Shin, E.A.; Shon, J.C.; Sun, H.; Kim, J.E.; Jung, J.W.; Lee, H.; Pinanga, Y.; Song, D.G.; et al. Crosstalk between TM4SF5 and GLUT8 regulates fructose metabolism in hepatic steatosis. Mol. Metab. 2022, 58, 101451.
- Qi, X.; Tester, R.F. Fructose, galactose and glucose-In health and disease. Clin. Nutr. ESPEN 2019, 33, 18–28.
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781.
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043.
- El Hiani, Y.; Egom, E.E.; Dong, X.P. mTOR signalling: Jack-of-all-trades. Biochem. Cell Biol. 2019, 97, 58–67.
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446.
- Han, J.; Wang, Y. mTORC1 signaling in hepatic lipid metabolism. Protein Cell 2018, 9, 145–151.
- Koundouros, N.; Blenis, J. Targeting mTOR in the Context of Diet and Whole-body Metabolism. Endocrinology 2022, 163, bqac041.
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309.
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194.
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e12.
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31.
- Lee, S.A.; Lee, S.Y.; Cho, I.H.; Oh, M.A.; Kang, E.S.; Kim, Y.B.; Seo, W.D.; Choi, S.; Nam, J.O.; Tamamori-Adachi, M.; et al. Tetraspanin TM4SF5 mediates loss of contact inhibition through epithelial-mesenchymal transition in human hepatocarcinoma. J. Clin. Investig. 2008, 118, 1354–1366.
- Ko, D.; Kim, E.; Shin, E.A.; Nam, S.H.; Yoon, J.; Lee, J.S.; Lee, Y.; Park, S.; Ha, K.; Choi, S.Y.; et al. Therapeutic effects of TM4SF5-targeting chimeric and humanized monoclonal antibodies in hepatocellular and colon cancer models. Mol. Ther. Oncolytics 2022, 24, 452–466.
- Suschek, C.V.; Schnorr, O.; Hemmrich, K.; Aust, O.; Klotz, L.O.; Sies, H.; Kolb-Bachofen, V. Critical role of L-arginine in endothelial cell survival during oxidative stress. Circulation 2003, 107, 2607–2614.
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383.
- Qiu, F.; Huang, J.; Sui, M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett. 2015, 364, 1–7.
- Lee, S.A.; Ryu, H.W.; Kim, Y.M.; Choi, S.; Lee, M.J.; Kwak, T.K.; Kim, H.J.; Cho, M.; Park, K.H.; Lee, J.W. Blockade of four-transmembrane L6 family member 5 (TM4SF5)-mediated tumorigenicity in hepatocytes by a synthetic chalcone derivative. Hepatology 2009, 49, 1316–1325.
- Ahn, H.-M.; Ryu, J.; Song, J.M.; Lee, Y.; Kim, H.-J.; Ko, D.; Choi, I.; Kim, S.J.; Lee, J.W.; Kim, S. Anti-cancer activity of novel TM4SF5-targeting antibodies through TM4SF5 neutralization and immune cell-mediated cytotoxicity. Theranostics 2017, 7, 594–613.
- Toribio, V.; Yanez-Mo, M. Tetraspanins interweave EV secretion, endosomal network dynamics and cellular metabolism. Eur. J. Cell Biol. 2022, 101, 151229.
- Busto, J.V.; Wedlich-Soldner, R. Integration Through Separation-The Role of Lateral Membrane Segregation in Nutrient Uptake. Front. Cell Dev. Biol. 2019, 7, 97.
- Cai, S.; Deng, Y.; Peng, H.; Shen, J. Role of Tetraspanins in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 723341.
- Lee, S.A.; Park, K.H.; Lee, J.W. Modulation of signaling between TM4SF5 and integrins in tumor microenvironment. Front. Biosci. 2011, 16, 1752–1758.
- Sala-Valdes, M.; Ailane, N.; Greco, C.; Rubinstein, E.; Boucheix, C. Targeting tetraspanins in cancer. Expert Opin. Ther. Targets 2012, 16, 985–997.
- Gu, J.; Geng, M.; Qi, M.; Wang, L.; Zhang, Y.; Gao, J. The role of lysosomal membrane proteins in glucose and lipid metabolism. FASEB J. 2021, 35, e21848.
- Rahmani, S.; Defferrari, M.S.; Wakarchuk, W.W.; Antonescu, C.N. Energetic adaptations: Metabolic control of endocytic membrane traffic. Traffic 2019, 20, 912–931.
- Murayama, Y.; Shinomura, Y.; Oritani, K.; Miyagawa, J.; Yoshida, H.; Nishida, M.; Katsube, F.; Shiraga, M.; Miyazaki, T.; Nakamoto, T.; et al. The tetraspanin CD9 modulates epidermal growth factor receptor signaling in cancer cells. J. Cell Physiol. 2008, 216, 135–143.
- Odintsova, E.; Voortman, J.; Gilbert, E.; Berditchevski, F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. J. Cell Sci. 2003, 116, 4557–4566.
- Huang, W.; Febbraio, M.; Silverstein, R.L. CD9 tetraspanin interacts with CD36 on the surface of macrophages: A possible regulatory influence on uptake of oxidized low density lipoprotein. PLoS ONE 2011, 6, e29092.
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47.
- Zhao, K.; Wang, Z.; Hackert, T.; Pitzer, C.; Zoller, M. Tspan8 and Tspan8/CD151 knockout mice unravel the contribution of tumor and host exosomes to tumor progression. J. Exp. Clin. Cancer Res. 2018, 37, 312.
- Srinivas, A.N.; Suresh, D.; Santhekadur, P.K.; Suvarna, D.; Kumar, D.P. Extracellular Vesicles as Inflammatory Drivers in NAFLD. Front. Immunol. 2021, 11, 627424.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
09 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No