 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manuela Oliveira | -- | 2687 | 2022-08-01 21:28:21 | | | |
| 2 | Rita Xu | Meta information modification | 2687 | 2022-08-02 02:53:15 | | | | |
| 3 | Manuela Oliveira | + 1 word(s) | 2688 | 2022-08-02 10:36:16 | | |
Video Upload Options
Fungi are amongst the most abundant and diverse organisms. Despite being widely known for their adverse role in food spoilage or as pathogens for humans, animals, or plants, they also present several beneficial effects. Fungi contribute to human well-being due to their role as decomposers, degrading decay matter into smaller molecules which can be easily used by other ecosystem members. These organisms can produce medicinal compounds or modulate protective immune responses in human intestine. Fungi intervene in diverse food processes or act as a food supply. Due to fungal diversity, the unequivocal identification of these organisms is crucial to increasing their practical applications and decreasing their adverse effects.
1. Introduction
2. DNA-Based Molecular Markers
2.1. Restriction Fragment Length Polymorphism (RFLP)
2.2. Random Amplification of Polymorphic DNA (RAPD)
2.3. Amplified Fragment Length Polymorphism (AFLP)
2.4. Inter-Simple Sequence Repeats (ISSR)
2.5. Variable Number of Tandem Repeats (VNTR)
2.6. Single-Nucleotide Polymorphisms (SNP)
2.7. Small Insertions or Deletions (InDels)
Insertions or Deletions are fragments of different sizes (ranging from 1 to 1000 bp) inserted or lost at a given location in the genome. These markers are very stable within the genome and can be relevant for population studies [39]. These markers are co-dominant, with high polymorphism and very abundant, presenting both high reliability and reproducibility.
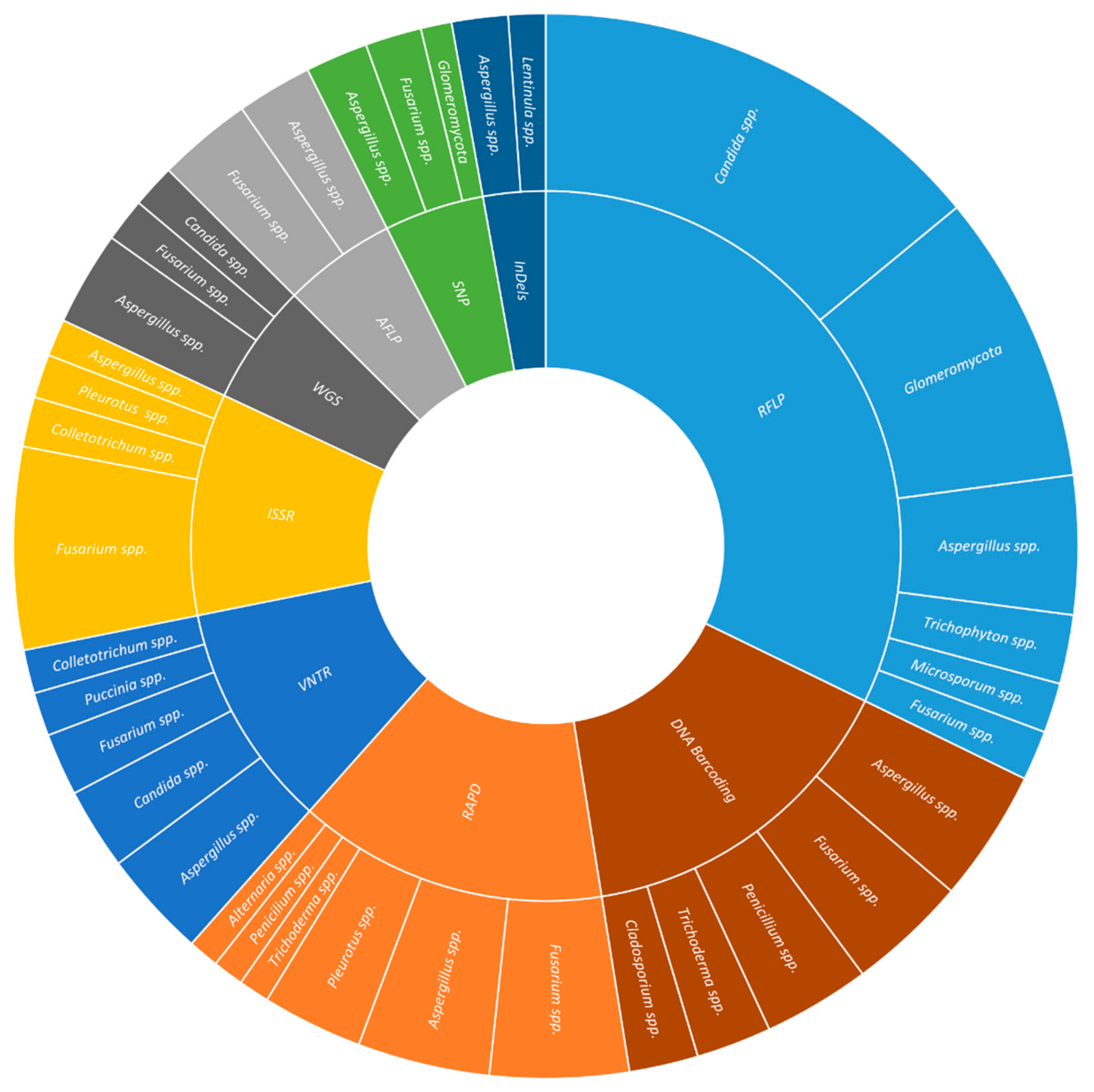
InDels have been used in studies of plant (29.0%) and human health (17.0%), and food and beverage (17.0%). The most commonly studied plant pathogens were Fusarium spp. (16.7%), Botrytis spp. (10.0%), and Puccinia spp. (10%), covering aspects such as phytopathogen genetic diversity (23.7%), the distinction between the species of the complex (7.9%), resistance to fungicides (7.9%), in crops like wheat (21.4%), oilseed rape (14.3%), and soybean (14.3%). The most frequently studied human pathogens were Cryptococcus spp. (19.2%), Aspergillus spp. (11.5%), and Trichophyton spp. (11.5%), elucidating aspects such as genetic diversity (40.0%), recombination (15.0%), and virulence factors (15.0%), associated to diseases as (25.0%), diarrhea (16.7%), and other enteric diseases (16.7%) (Figure 1).
2.8. DNA Barcoding
DNA barcoding relies on the use of a single universal marker - DNA barcode - for the rapid and efficient identification and classification of species by non-specialists [40], similar to the Universal Product Code that identifies products in a supermarket [40].
Each Kingdom possesses a particular barcode gene or genes [41][42]. Fungal ITS is frequently proposed as the first universal barcoding marker. However, this barcode does not provide sufficient resolution being common to use secondary barcodes [i.e., intergenic spacer (IGS), β-tubulin II (TUB2), DNA-directed RNA polymerase II largest (RPB1) and second largest (RPB2) subunits, translational elongation factor 1α (TEF-1α), DNA topoisomerase I (TOP1), phosphoglycerate kinase (PGK), and cytochrome c oxidase subunit I (COX1) and subunit II (COX2), 28S nrDNA (LSU), and 18S nrDNA (SSU)] [43][44].
These markers are co-dominant and present high genomic abundance, being their analysis highly reliable and reproducible. A disadvantage is that the same marker cannot be universally used for all fungal species, and prior knowledge of the DNA sequence is required [45].
DNA barcoding has been used in studies on plant (21.4%) and human health (13.4%), and food and beverage (12.5%). In plant health, the most used barcode genes were ITS (36.2%), TEF-1α (13.8%), and LSU (12.1%), applied to Fusarium spp. (11.6%), Alternaria spp. (2.9%), Curvularia spp. (2.9%), Diaporthe spp. (2.9%), Exserohilum spp. (2.9%), Mycosphaerella spp. (2.9%), and Neofusicoccum spp. (2.9%) collected from several crops (e.g., blueberry, chili, sorghum, Pinus). The aims of such studies were diagnosis (15.8%), identification of leaf spot pathogens (15.8%), and quarantine species (10.5%). In human health, the most common barcode genes were ITS (33.3%), β-TUB (18.5%), and TEF-1α (14.8%). The human pathogens included were Scedosporium spp. (9.7%), Aspergillus spp. (6.5%), Cunninghamella spp. (6.5%), and Fusarium spp. (6.5%); whereas, the major diseases were invasive fungal infections (28.6%), keratitis (21.4%), and opportunistic fungi (14.3%). In food and beverage production and spoilage, the barcodes were mainly ITS (43.3%), LSU (20.0%), and β-TUB (10.0%), applied to the study of Lactarius spp. (2.7%), Penicillium spp. (2.7%), and Pleurotus spp. (2.7%) associated either with edible mushrooms (53.3%) or dairy products (20.0%) (Figure 1)
2.9. Massive Parallel Sequencing (MPS)
The development of Massive Parallel Sequencing (MPS) techniques allow for a deeper knowledge of the entire microbial composition of a given environment [46] or determining the complete genome sequence of a single microorganism [47].
MPS advantages include its high-throughput capacity; a single protocol can be applied for the identification and genotyping of all microorganisms; no need for DNA cloning; no need for an a priori knowledge about the sequence of a particular gene/genome; no need for isolation and culture of the microorganism to be studied; and reduced costs and the turnaround time [48][49]. As disadvantages, these techniques are associated with high requirements for data storage and the biases introduced by each step of the protocol [50].
Metabarcoding is the automated identification of various organisms present in a single bulk sample or from an environmental sample with degraded DNA using a species-specific genetic marker (DNA meta-barcode). Metabarcoding studies included plant health (14.9%), food and beverage (14.4%), and soil (10.9%). In plant health studies, the most frequently used metabarcodes were ITS2 (31.0%) and ITS1 (27.6%) either isolated or in a combination of both (13.8%). The selected works included studies on fungal community (30.8%), endophytes (26.9%), trunk disease (11.5%) applied to forest trees (27.3%), cereals (22.7%), and fruit trees (13.6%). In food and beverage production and spoilage, the same metabarcodes were used (ITS2: 36.0%; ITS1: 16.0%; ITS1+ITS2: 12.0%). Sequencing was used for the identification of species involved in beverage fermentation (26.9%), food contamination (19.2%), and the production of dairy products (15.4%). In the soil’s fungal diversity studies, the three usual metabarcodes were used (ITS2: 20.0%; ITS1: 15.0%; ITS1+ITS2: 15.0%). The selected soils were from forests (21.1%), agro-environment (15.8%), and public gardens (15.8%) (Figure 1).
Whole genome sequencing (WGS) includes two different techniques: de novo genome assembly (the species to be studied has not been previously sequenced and assembled) and re-sequencing, which identifies genome-wide variants comparing an existing reference assembly with a sequenced isolate through the alignment of sequence reads against the reference [47].
WGS has been used to elucidate aspects associated with plant (32.9%) and human health (13.4%), and food and beverage (12.8%). In plant health, the main fungal phytopathogens studied were Fusarium spp. (12.1%), Trichoderma spp. (6.1%), and Venturia spp. (6.1%); while the major diseases analyzed were rot (27.8%), blight (19.4%), and cancer (8.3%). The most common studied cultures were soybean (13.6%), grapevine (9.1%), sugarcane (9.1%), and wheat (9.1%). In human health, the most common fungal species were Candida spp. (42.1%), the etiological agent of candidiasis and candidemia (42.1%), and Aspergillus spp. (15.8%) causing aspergillosis (10.5%) and other infections (5.3%). In food and beverages production and spoilage, species such as Agaricus bisporus (4.8%), Auricularia spp. (4.8%), Flammulina spp. (4.8%), were studied for their interest as edible mushroom (55.0%); while Aspergillus spp. (23.8%) and Penicillium spp. (19.0%) were studied for their role in food spoilage (45.0%) (Figures 1).
3. Conclusions
Identification of fungal species is a crucial aspect of many fields in science. Mainly associated with plant, animal, and human health or food production and spoilage. Throughout the years, the scientific community has developed several molecular techniques to obtain information on these species. Nowadays, these markers range from non-PCR, PCR-based techniques to more advanced MPS-based techniques
References
- Schmit, J.P.; Mueller, G.M. An estimate of the lower limit of global fungal diversity. Biodivers. Conserv. 2007, 16, 99–111.
- Blackwell, M. The Fungi: 1, 2, 3… 5.1 million species? Am. J. Bot. 2011, 98, 426–438.
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10.
- Vu, D.; Groenewald, M.; de Vries, M.; Gehrmann, T.; Stielow, B.; Eberhardt, U.; Al-Hatmi, A.; Groenewald, J.Z.; Cardinali, G.; Houbraken, J.; et al. Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud. Mycol. 2018, 91, 23–36.
- Adnan, M.; Islam, W.; Gang, L.; Chen, H.Y. Advanced research tools for fungal diversity and its impact on forest ecosystem. Environ. Sci. Pollut. Res. 2022, 29, 45044–45062.
- Barros, J.; Seena, S. Fungi in Freshwaters: Prioritising Aquatic Hyphomycetes in Conservation Goals. Water 2022, 14, 605.
- Gonçalves, M.F.; Esteves, A.C.; Alves, A. Marine Fungi: Opportunities and Challenges. Encyclopedia 2022, 2, 559–577.
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638.
- Shamim, M.; Kumar, P.; Kumar, R.R.; Kumar, M.; Kumar, R.R.; Singh, K.N. Assessing Fungal Biodiversity Using Molecular Markers. In Molecular Markers in Mycology; Fungal Biology; Singh, B.P., Gupta, V.K., Eds.; Springer: Cham, Switzerland, 2017; pp. 305–333.
- Gautam, A.K.; Verma, R.K.; Avasthi, S.; Bohra, Y.; Devadatha, B.; Niranjan, M.; Suwannarach, N. Current insight into traditional and modern methods in fungal diversity estimates. J. Fungi 2022, 8, 226.
- Tahir, A.; Iqbal, I.; Talib, K.M.; Luhuai, J.; Chen, X.; Akbar, A.; Asghar, A.; Ali, I. Modern Tools for the Identification of Fungi, Including Yeasts. In Extremophilic Fungi; Sahay, S., Ed.; Springer: Singapore, 2022; pp. 33–51.
- Chaudhary, R.; Kumar, G.M. Restriction fragment length polymorphism. In Encyclopedia of Animal Cognition and Behavior; Vonk, J., Shackelford, T.K., Eds.; Springer: Cham, Switzerland, 2019.
- Atoui, A.; El Khoury, A. PCR-RFLP for Aspergillus species. In Mycotoxigenic Fungi. Methods in Molecular Biology; Moretti, A., Susca, A., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1542, pp. 313–320.
- Kennedy, N.; Clipson, N. Fingerprinting the fungal community. Mycologist 2003, 17, 158–164.
- Gryta, A.; Frąc, M. Methodological aspects of multiplex terminal restriction fragment length polymorphism-technique to describe the genetic diversity of soil bacteria, archaea and fungi. Sensors 2020, 20, 3292.
- Welsh, J.; McClelland, M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990, 18, 7213–7218.
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535.
- Bardakci, F. Random amplified polymorphic DNA (RAPD) markers. Turk. J. Biol. 2001, 25, 185–196.
- Matthes, M.C.; Daly, A.; Edwards, K.J. Amplified fragment length polymorphism (AFLP). In Molecular Tools for Screening Biodiversity; Karp, A., Isaac, P.G., Ingram, D.S., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 183–190.
- Blears, M.; de Grandis, S.; Lee, H.; Trevors, J.T. Amplified fragment length polymorphism (AFLP): A review of the procedure and its applications. J. Ind. Microbiol. Biotech. 1998, 21, 99–114.
- Savelkoul, P.H.M.; Aarts, H.J.M.; de Haas, J.; Dijkshoorn, L.; Duim, B.; Otsen, M.; Rademaker, J.L.; Schouls, W.L.; Lenstra, J.A. Amplified-fragment length polymorphism analysis: The state of an art. J. Clin. Microbiol. 1999, 37, 3083–3091.
- Fry, N.K.; Savelkoul, P.H.; Visca, P. Amplified fragment length polymorphism analysis. In Molecular Epidemiology of Microorganisms. Methods in Molecular Biology; Caugant, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 551, pp. 89–104.
- Zietkiewicz, E.; Rafalski, A.; Labuda, D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 1994, 20, 176–183.
- Hassel, K.; Gunnarsson, U. The use of inter simple sequence repeats (ISSR) in bryophyte population studies. Lindbergia 2003, 28, 152–157.
- Jeffreys, A.; Wilson, V.; Thein, S. Hypervariable ‘minisatellite’ regions in human DNA. Nature 1985, 314, 67–73.
- Jeffreys, A.J.; Neumann, R.; Wilson, V. Repeat unit sequence variation in minisatellites: A novel source of DNA polymorphism for studying variation and mutation by single molecule analysis. Cell 1990, 60, 473–485.
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397.
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465.
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611.
- Fischer, M.C.; Rellstab, C.; Leuzinger, M.; Roumet, M.; Gugerli, F.; Shimizu, K.K.; Holderegger, R.; Widmer, A. Estimating genomic diversity and population differentiation–an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genom. 2017, 18, 1–15.
- Tsykun, T.; Rellstab, C.; Dutech, C.; Sipos, G.; Prospero, S. Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 2017, 119, 371–380.
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genom. 2020, 21, 382.
- Estoup, A.; Jarne, P.; Cornuet, J.M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002, 11, 1591–1604.
- Lander, E.S. The new genomics: Global views of biology. Science 1996, 274, 536–539.
- Zhao, Z.; Fu, Y.-X.; Hewett-Emmett, D.; Boerwinkle, E. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 2003, 312, 207–213.
- Van der Heyden, H.; Dutilleul, P.; Brodeur, L.; Carisse, O. Spatial distribution of single-nucleotide polymorphisms related to fungicide resistance and implications for sampling. Phytopathology 2014, 104, 604–613.
- Kaiser, S.A.; Taylor, S.A.; Chen, N.; Sillett, T.S.; Bondra, E.R.; Webster, M.S. A comparative assessment of SNP and microsatellite markers for assigning parentage in a socially monogamous bird. Mol. Ecol. Resour. 2017, 17, 183–193.
- Dutech, C.; Enjalbert, J.; Fournier, E.; Delmotte, F.; Barres, B.; Carlier, J.; Tharreau, D.; Giraud, T. Challenges of microsatellite isolation in fungi. Fungal Genet. Biol. 2007, 44, 933–949.
- Yang, J.; He, J.; Wang, D.; Shi, E.; Yang, W.; Geng, Q.; Wang, Z. Progress in research and application of InDel markers. Biodivers. Sci. 2016, 24, 237–243.
- Frézal, L.; Leblois, R. Four years of DNA barcoding: Current advances and prospects. Infect. Genet. Evol. 2008, 8, 727–736.
- Lebonah, D.E.; Dileep, A.; Chandrasekhar, K.; Sreevani, S.; Sreedevi, B.; Pramoda Kumari, J. DNA barcoding on bacteria: A review. Adv. Biol. 2014, 2014, 541787.
- Kress, W.J.; Erickson, D.L. DNA barcodes: Methods and protocols. Methods Mol. Biol. 2012, 858, 3–8.
- Valentini, A.; Pompanon, F.; Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol. 2009, 24, 110–117.
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645.
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246.
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14.
- Xu, J. Fungal DNA barcoding. Genome 2016, 1, 913–932.
- Mbareche, H.; Dumont-Leblond, N.; Bilodeau, G.J.; Duchaine, C. An overview of bioinformatics tools for DNA meta-barcoding analysis of microbial communities of bioaerosols: Digest for microbiologists. Life 2020, 10, 185.
- Cuomo, C.A. Harnessing Whole Genome Sequencing in Medical Mycology. Curr. Fungal Infect. Rep. 2017, 11, 52–59.
- Oliveira, M.; Amorim, A. Microbial forensics: New breakthroughs and future prospects. Appl. Microbiol. Biotechnol. 2018, 102, 10377–10391.

