Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martin H Steinberg | -- | 1837 | 2022-07-29 15:31:41 | | | |
| 2 | Peter Tang | Meta information modification | 1837 | 2022-08-01 02:59:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies. Encyclopedia. Available online: https://encyclopedia.pub/entry/25672 (accessed on 09 August 2026).
Steinberg MH. Fetal Hemoglobin in Sickle Hemoglobinopathies. Encyclopedia. Available at: https://encyclopedia.pub/entry/25672. Accessed August 09, 2026.
Steinberg, Martin H.. "Fetal Hemoglobin in Sickle Hemoglobinopathies" Encyclopedia, https://encyclopedia.pub/entry/25672 (accessed August 09, 2026).
Steinberg, M.H. (2022, July 29). Fetal Hemoglobin in Sickle Hemoglobinopathies. In Encyclopedia. https://encyclopedia.pub/entry/25672
Steinberg, Martin H.. "Fetal Hemoglobin in Sickle Hemoglobinopathies." Encyclopedia. Web. 29 July, 2022.
Copy Citation
Fetal hemoglobin (HbF; α2γ2), encoded by two nearly identical γ-globin genes (HBG2, HBG1) that are part of the β-globin gene (HBB) cluster (11p15.4), comprises 70 to 90% of the hemolysate in newborns, falling to <1% after 12 months. HbF usually consists of 4 to 10% of total hemoglobin in adults of African descent with sickle cell anemia. Rarely, their HbF levels reach more than 30%. High HbF levels are sometimes a result of β-globin gene deletions or point mutations in the promoters of the HbF genes.
hereditary persistence of fetal hemoglobin
gamma-globin gene
gene deletion
globin gene expression
genotype/phenotype
1. Fetal Hemoglobin Levels in Sickle Cell Anemia
Fetal hemoglobin (HbF; α2γ2), encoded by two nearly identical γ-globin genes (HBG2, HBG1) that are part of the β-globin gene (HBB) cluster (11p15.4), comprises 70 to 90% of the hemolysate in newborns, falling to <1% after 12 months [1]. The “switch” from fetal to adult hemoglobin synthesis in sickle cell anemia (homozygosity for the sickle hemoglobin gene) takes 5 to 10 years and is rarely complete. The sickle hemoglobin gene (HbS) is found on five common genetic backgrounds, or haplotypes. Patients of African descent have Bantu, Benin, Cameroon or Senegal haplotypes; in most of these patients a stable HbF level of ~4 to 10% is reached at ~5 years of age. In individuals of Middle Eastern or Indian extract with the Arab–Indian haplotype, stable HbF levels of ~17% are achieved at ~10 years of age [2][3]. Sometimes, in treatment-naive adults of African descent HbF levels between 15 and 30% are present; this can be accompanied by mild disease [4]. Patients with the Arab–Indian haplotype can have HbF far above 20%, but no informative genetic studies of the very high HbF phenotype have been done in this population; the following sections deal only with patients of African ancestry [5].
2. Persistent High HbF: Mutations of the β-globin Gene Cluster
2.1. Hereditary Persistence of Fetal Hemoglobin and δβ Thalassemia
Hereditary persistence of fetal hemoglobin, or HPFH, a term perhaps first coined in 1958, is defined by deletions or point mutations within the HBB gene cluster [6]. Large deletions that result in higher than normal levels of HbF persisting into adulthood cause the most frequently recognized type of HPFH (Figure 1). δβ thalassemia is also caused by HBB gene cluster deletions but has some hematologic features of thalassemia. HPFH and δβ thalassemia are overlapping phenotypes; their distinction is historical and semantic. The sizes of the causative deletions overlap while their 5′ and 3′ breakpoints differ. Deletions of different sizes with diverse breakpoints might differentially change the interactions among γ-globin genes and their upstream and downstream regulatory elements, such as the locus control region (LCR), and transcription factor complexes. This could account for the wide variation in HbF levels in heterozygous carriers of these mutations and in compound heterozygotes with the HbS gene. Identical HPFH-causing mutations, can be associated with dissimilar HbF levels. Although the causes of this are unknown, this might be a result of the inheritance of the minor alleles of the quantitative trait loci (QTL) modulating HbF production that are associated with higher HbF and are discussed below. Reports of QTL genotypes in HPFH have not been published.
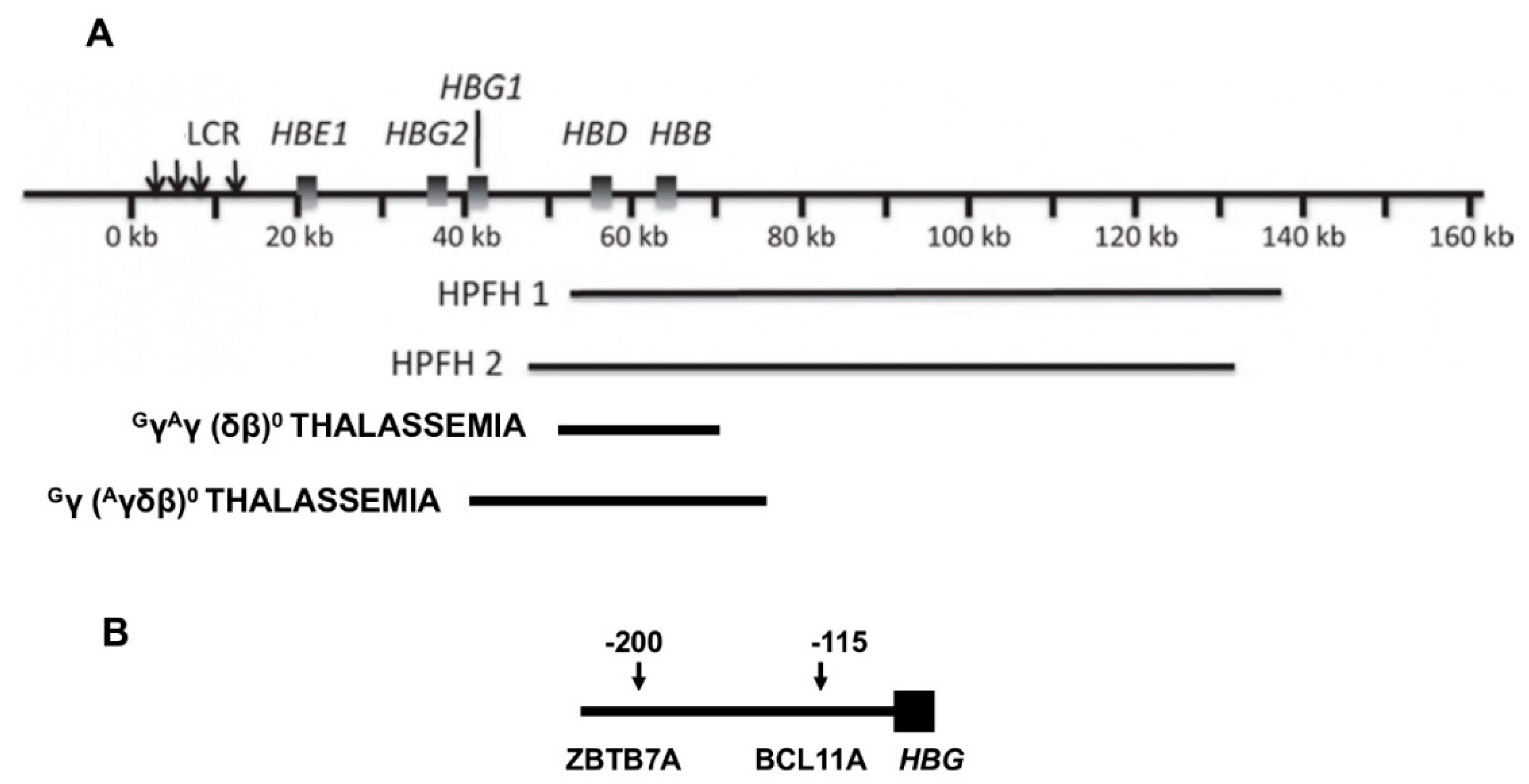
Figure 1. Deletions in the HBB gene cluster associated with increased HbF. (A) Displayed are the genes in the HBB gene cluster. The locus control region (LCR) that is upstream of HBE forms contacts with the promoters of the genes in this cluster affecting their transcription. When HBB and HBD are deleted, interactions between the promoters of the γ-globin genes and the LCR are favored. Beneath the HBB gene cluster are shown the locations and approximate sizes of common HPFH and δβ thalassemia deletions. (B) At positions −115 and −200 upstream of both HBG2 and HBG1 are binding motifs for the γ-globin gene repressors, ZBTB7A at −200 bp and BCL11A at −115 bp, respectively. Point mutations and small deletions in these regions and in nearby sites in the promoters, cause HPFH by altering the binding of these and other transcription factors.
2.2. HbS-Gene Deletion HPFH and δβ Thalassemia
Deletions of about 85 kilobases that remove HBB and HBD account for the most commonly reported HPFH 1 and HPFH 2 mutations although many other HPFH deletions have been recognized [7]. In HPFH 1 and 2, HBG2 and HBG1 are preserved and HbF levels of ~30% are present in heterozygotes; homozygotes have 100% HbF. Based on hematologic and electrophoretic findings, about 0.1% of African Americans were estimated to have the HPFH genotype; an estimation of 1 case in 14,000 was made from the Jamaican birth cohort [8][9][10]. The nearly uniform presence of mild microcytosis suggests that compensatory increase in γ-globin chain (HBG2/HBG1) production is nearly, but not totally, complete even though balanced α- to non-α-globin synthesis has been found [11]. All erythrocytes contain some HbF. This pancellular distribution was first pointed out by Conley in 1963, who attributed the benignity of HbS-HPFH to this HbF distribution [9]. When HbS is present in compound heterozygosity with an HPFH deletion, HbS polymerization is inhibited in most erythrocytes resulting in nearly normal hematology; 51Cr red cell half-life was normal [9][12]. A problem in understanding the phenotype of HbS-HPFH is that the hemoglobin genotypes are not always precisely defined. In the largest series of molecularly defined HbS-HPFH, where gap-PCR assays were designed specifically to detect HPFH 1 and HPFH 2, HbF was 31.3 ± 2.4% in 30 cases aged >5 year [12].
2.3. HbS-HPFH Due to Point Mutations
This genotype, the result of many different mutations in the promoters of HBG2 and HBG1 and a single C > T polymorphism in HBG2, is associated with a diversity of HbF levels. The point mutation can be in cis or in trans to the HbS gene [13]. Once referred to as heterocellular HPFH, HbF can be distributed both pancellularly in cases where HbF levels are high or heterocellularly when HbF is lower, a likely reflection of the level of HBG2 or HBG1 expression [4]. Promoter mutations and the HBG2 polymorphism (rs7482144) increase HbF as a result of altered binding of the major HbF repressors, ZBTB7A and BCL11A, to their binding motifs and perhaps through the recruitment of other transcriptional activators like GATA1 to their nearby binding motifs. The most important HbF suppressor motifs lie ~200 and ~115 base-pairs upstream of the transcriptional start sites of each γ-globin gene and bind ZBTB7A and BCL11A, respectively [14][15]. Disruption of the HBG2 QTL 158 bps upstream of this gene and marked by rs7482144, increased HbF about half as much as disruption of the ZBTB7A and BCL11A binding motifs. Rs7482144 affects only HBG2, whereas both γ-globin genes are modulated by ZBTB7A and BCL11A, perhaps accounting for this difference [16][17]. The diversity of HbF levels associated with point mutations at the same nucleotide or adjacent nucleotides might be explained by how the physical interactions between the transcription factor and its binding motifs or interactions among the proteins in a transcription factor complex are perturbed.
3. Quantitative Trait Loci Modulating HbF Production
Most patients with HbS and very high levels of HbF do not have any of the aforementioned HPFH mutations and continue to have hemolytic anemia and sickle vasoocclusive complications, albeit perhaps at a lower rate. This is likely to be a result of the extreme heterogeneity of HbF distribution amongst F-cells, where despite the high total HbF level, a clinically important fraction of erythrocytes contain insufficient HbF to protect them from HbS polymer inflicted damage [4]. Perhaps interpatient variability in erythropoietic stress accounts for very high HbF levels in some individuals lacking HPFH mutations. Studies in primates suggested wide inter-animal differences in the HbF response to stress erythropoiesis [18].
Recent reviews have summarized the molecular basis of HbF repression in the fetus and the ensuing switch to adult hemoglobins [19][20][21]. Higher than expected levels of HbF in sickle cell anemia can be a result of polymorphisms of the major QTL that modulate HbF gene expression. Three QTL, BCL11A, MYB and HBG2, are known to be associated with HbF levels accounting for 10 to 50% of HbF variance [22]. The minor allele frequencies of the sentinel variants, or single nucleotide polymorphisms (SNPs) of these QTL, in sickle cell anemia varies by geographic origin. In sickle cell anemia, the sentinel SNP of the HBG2 QTL, rs7482144 is present only in the Senegal haplotype of the HbS gene common in patients from West Central Africa and in patients who have the Arab-Indian HbS haplotype and ancestry from Eastern Saudi Arabia or India.
From the records of the Hemoglobin Diagnostic Reference Laboratory (www.bu.edu/sicklecell), Akinsheye and her coworkers culled samples from 20 African Americans who were homozygous for the HbS gene with HbF of 17.2 ± 4.8 g/dL, were more than five years old, had MCV < 100 fL and who had neither gene deletion HPFH nor point mutations in HbF gene promoters. These individuals were compared with 30 similar patients with HbF of 5.0 ± 2.5% [23]. Minor alleles of the sentinel SNPs of BCL11A (rs766432) and MYB (rs9399137) were more frequent in the high HbF group and accounted for 20% of HbF variance. The number of minor alleles present (NOMAP score) [24] in the high HbF patients was significantly greater than in the low HbF controls. These results were replicated when 56 HbS homozygotes with HbF of 20.7 ± 8.2%, who were followed in the Cooperative Study of Sickle Cell Disease (CSSCD), were compared with 489 individuals with HbF of 3.1 ± 1.5%. The minor allele frequencies of BCL11A, MYB and HBG2 were all significantly higher in the high HbF group. Primate studies have suggested that erythropoietic stress increases γ-globin gene expression when BCL11A was downregulated using CRISPR-Cas9 editing of its erythroid enhancer [25].
4. Therapeutic Targeting HbF QTL and Mutations
Understanding the mechanisms of some instances of unusually high HbF in sickle cell anemia and knowing the clinical benefits associated with high pancellular levels of HbF is being exploited for possible therapeutic benefit.
The ~13 kb Sicilian HPFH deletion was created in CD34+ hematopoietic progenitor cells using CRISPR-Cas9 in an attempt to reproduce the high HbF associated with this deletion [26]. In erythroid colonies derived from cells homozygous for this deletion, there was no HBB mRNA; cells heterozygous for the deletion synthesized half as much HBB mRNA as control cells. The ratio of HBG/HBB+HBG mRNA was ~0.1 in controls, ~0.3 in deletion heterozygotes and 1 in deletion homozygotes. Based on these studies, to be applicable therapeutically, only cells heterozygous for the deletion are needed.
CRISPR-Cas9 was used to mutate the 13 bp sequence in the promoters of HBG2 and HBG1 that is associated with HPFH. Edited progenitors derived from HUDEP-2 cells expressing HbA gave rise to ~50% F-cells and increased the HBG/HBB+HBG mRNA ratio; 58% of the erythroid cells derived from edited CD34+ progenitors were F-cells; HbF was ~20%. Erythrocytes derived from sickle CD34+ progenitors were 90% F-cells and protected from hypoxia-induced sickling [16][27].
CRISPR-Cas9 editing in both HUDEP-2 cells and CD34+ progenitors from patients with sickle cell anemia generated small deletions disrupting the binding motifs for: ZBTB7A 195–197 bp upstream of both HBG2 and HBG1; the BCL11A binding motif 115 bp upstream of both HBG2 and HBG1; and the QTL 158 bp upstream of HBG2. These induced mutations mimicked HPFH mutations. The editing efficiency was >80%. Editing the ZBTB7A binding site in patient CD34+ cells resulted in: γ-globin transcripts forming ~50% of all non-α transcripts; ~80 F-cells; HbF comprising ~50% of hemoglobin tetramers; and ~70 of cells protected from hypoxia-induced sickling in vitro [16].
References
- Nagel, R.L.; Steinberg, M.H. Hemoglobins of the embryo and fetus and minor hemoglobins of adults. In Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management, 1st ed.; Chapter 10; Steinberg, M.H., Forget, B.G., Higgs, D.R., Nagel, R.L., Eds.; Cambridge University Press: Cambridge, UK, 2001.
- Solovieff, N.; Milton, J.N.; Hartley, S.W.; Sherva, R.; Sebastiani, P.; Dworkis, D.A.; Klings, E.S.; Farrer, L.A.; Garrett, M.E.; Ashley-Koch, A.; et al. Fetal hemoglobin in sickle cell anemia: Genome-wide association studies suggest a regulatory region in the 5’ olfactory receptor gene cluster. Blood 2010, 115, 1815–1822.
- Ngo, D.; Bae, H.; Steinberg, M.H.; Sebastiani, P.; Solovieff, N.; Baldwin, C.T.; Melista, E.; Safaya, S.; Farrer, L.A.; Al-Suliman, A.M.; et al. Fetal hemoglobin in sickle cell anemia: Genetic studies of the Arab-Indian haplotype. Blood Cells Mol. Dis. 2013, 51, 22–26.
- Steinberg, M.H.; Chui, D.H.; Dover, G.J.; Sebastiani, P.; Alsultan, A. Fetal hemoglobin in sickle cell anemia: A glass half full? Blood 2014, 123, 481–495.
- Al-Ali, A.K.; Alsulaiman, A.; Alzahrani, A.J.; Obeid, O.T.; Vatte, C.B.; Cyrus, C.; Alnafie, A.N.; Alali, R.A.; Alfarhan, M.; Mozeleski, B.; et al. Prevalence and diversity of haplotypes of sickle cell disease in the Eastern Province of Saudi Arabia. Hemoglobin 2020, 1–4.
- Jacob, G.F.; Raper, A.B. Hereditary persistence of foetal haemoglobin production, and its interactions with sickle-cell trait. Br. J. Haematol. 1958, 4, 138–149.
- Wood, W.G. Hereditary persistence of fetal hemoglobin and δβ thalassemia. In Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management, 1st ed.; Chapter 15; Steinberg, M.H., Forget, B.G., Higgs, D.R., Nagel, R.L., Eds.; Cambridge University Press: Cambridge, UK, 2001.
- Bradley, T.B., Jr.; Brawner, J.N.; Conley, C.L. Further observations on an inherited anomaly characterized by persistence of fetal hemoglobin. Bull. Johns Hopkins Hosp. 1961, 108, 242–257.
- Conley, C.L.; Weatherall, D.J.; Richardson, S.N.; Shepard, M.K.; Charache, S. Hereditary persistence of fetal hemoglobin: A study of 79 affected persons in 15 Negro families in Baltimore. Blood 1963, 21, 261–281.
- Serjeant, G.R.; Serjeant, B.E.; Forbes, M.; Haye, R.J.; Higgs, D.R.; Lehmann, H. Haemoglobin gene frequencies in the Jamaican population: A study of 100,000 newborns. Br. J. HaernatoI. 1986, 64, 253–262.
- Natta, C.L.; Niazi, G.A.; Ford, S.; Bank, A. Balanced globin chain synthesis in hereditary persistence of fetal hemoglobin. J. Clin. Investig. 1974, 54, 433–438.
- Ngo, D.A.; Aygun, B.; Akinsheye, I.; Hankins, J.S.; Bhan, I.; Luo, H.Y.; Steinberg, M.H.; Chui, D.H. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br. J. Haematol. 2012, 156, 259–264.
- Milner, P.F.; Leibfarth, J.D.; Ford, J.; Barton, B.P.; Grenett, H.E.; Garver, F.A. Increased HbF in sickle cell anemia is determined by a factor linked to the beta S gene from one parent. Blood 1984, 63, 64–72.
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 2018, 173, 430–442.
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.; Funnell, A.P.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503.
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Meneghini, V.; Masson, C.; De Cian, A.; Chalumeau, A.; et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020, 6, eaay9392.
- Shaikho, E.M.; Farrell, J.J.; Chui, D.H.K.; Sebastiani, P.; Steinberg, M.H. Cis- and trans-acting expression quantitative trait loci differentially regulate gamma-globin gene expression. BioRxiv 2018, 304899.
- DeSimone, J.; Biel, S.I.; Heller, P. Stimulation of fetal hemoglobin synthesis in baboons by hemolysis hypoxia. Proc. Natl. Acad. Sci. USA 1978, 75, 2937–2940.
- Vinjamur, D.S.; Bauer, D.E.; Orkin, S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018, 180, 630–643.
- Wienert, B.; Martyn, G.E.; Funnell, A.P.W.; Quinlan, K.G.R.; Crossley, M. Wake-up sleepy gene: Reactivating fetal globin for beta-hemoglobinopathies. Trends Genet. 2018, 34, 927–940.
- Orkin, S.H.; Bauer, D.E. Emerging genetic therapy for sickle cell disease. Ann. Rev. Med. 2019, 70, 257–271.
- Gardner, K.; Fulford, T.; Silver, N.; Rooks, H.; Angelis, N.; Allman, M.; Nkya, S.; Makani, J.; Howard, J.; Kesse-Adu, R.; et al. g(HbF): A genetic model of fetal hemoglobin in sickle cell disease. Blood Adv. 2018, 2, 235–239.
- Akinsheye, I.; Solovieff, N.; Ngo, D.; Malek, A.; Sebastiani, P.; Steinberg, M.H.; Chui, D.H. Fetal hemoglobin in sickle cell anemia: Molecular characterization of the unusually high fetal hemoglobin phenotype in African Americans. Am. J. Hematol. 2012, 87, 217–219.
- Galanello, R.; Sanna, S.; Perseu, L.; Sollaino, M.C.; Satta, S.; Lai, M.E.; Barella, S.; Uda, M.; Usala, G.; Abecasis, G.R.; et al. Amelioration of Sardinian β0 thalassemia by genetic modifiers. Blood 2009, 114, 3935–3937.
- Demirci, S.; Zeng, J.; Wu, Y.; Uchida, N.; Shen, A.H.; Pellin, D.; Gamer, J.; Yapundich, M.; Drysdale, C.; Bonanno, J.; et al. BCL11A enhancer edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Investig. 2020, 8, 140189.
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665.
- Traxler, E.A.; Yao, Y.; Wang, Y.D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016, 22, 987–990.
More
Information
Subjects:
Hematology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
01 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No