1. Translational Arrest Is Regulated by ERdj1 and ERdj2
ERdj1 is a transmembrane protein with its N-terminus in the ER lumen and its C-terminus in the cytosol
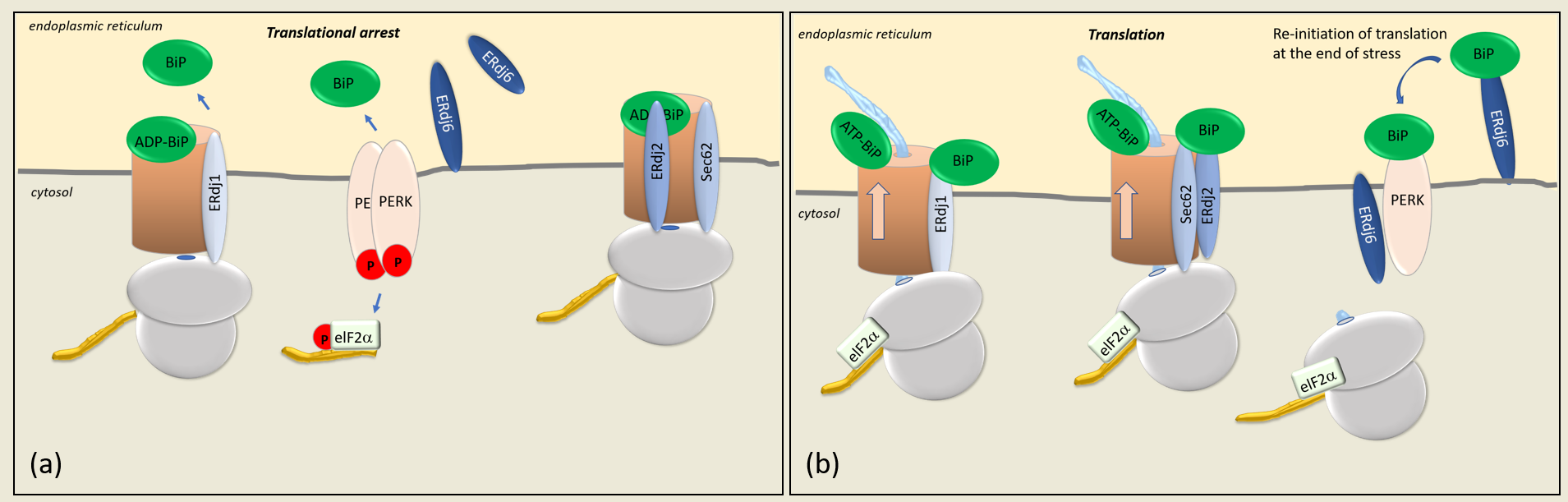
[1][2]. ERdj1 inhibits protein synthesis at the Sec61 translocon in a BiP- and ATP-dependent way (
Figure 1a). In vitro translation assays using preprolactin and luciferase as substrates suggested that the inhibition of protein synthesis is achieved by the first 21 N-terminal amino acids of the cytosolic domain of ERdj1, which bind to ribosomes close to the tunnel exit and to proteins emerging at the tunnel exit
[1][3][4][5]. Translation is arrested at its initiation, since ERdj1 is not able to inhibit the translation of preprolactin when added after translational initiation
[4]. The inhibitory function of ERdj1, as well as ribosome binding, seems to be mediated by a four-amino acid RKKR within the RKKRERKKK stretch
[4]. This polybasic amino acid stretch is similar to an amino acid stretch within the SRP, which also inhibits elongation
[3][6].
Figure 1. (a) At the translocon (brown cylinder), the pore is gated by BiP, which, in its ADP-bound form, closes the gate and opens it in its ATP-bound form. Translational arrest occurs when BiP dissociates from the luminal domains of ERdj1, ERdj2, and PERK. The release of BiP from PERK results in the dimerization of PERK, its autophosphorylation, and the subsequent phosphorylation of eIF2α (p-eIF2α), which inhibits eIF2α-dependent translation. (b) The translation of proteins is controlled by the co-chaperones ERdj1, ERdj2/Sec62, and ERdj6. In their BiP-bound forms, ERdj1 and ERdj2/Sec62 enable protein synthesis. The silencing of PERK and the PERK-signaling pathway occur by the binding of BiP to its luminal domain, which results in the release of eIF2α-dependent translation and protein synthesis. BiP, immunoglobin binding protein; PERK, protein kinase RNA-like endoplasmic reticulum kinase; eIF2α, alpha subunit of eukaryotic initiation factor 2.
In the presence of ERdj1, an increased amount of BiP associates with the ribosomal fractions, suggesting that ERdj1 plays a role in recruiting BiP to the ribosome/ERdj1 complex
[1][4]. A BiP mutant (BiP R197H), which is unable to bind to J-domains and experiments with the J-domain-deleted ERdj1 protein, revealed that the recruitment of BiP to the ERdj1–ribosome complex is ATP-dependent and requires the functional interaction of BiP and the ATPase-stimulating J-domain
[4]. The association of ERdj1 with the emerging protein and, accordingly, the translational arrest was shown to occur only in the absence of BiP
[4] (
Figure 1a). Upon the binding of BiP to ERdj1, the inhibition of protein translation by ERdj1 is released
[1][4] (
Figure 1b). ERdj1 thus seems to relay the information about luminal BiP availability to the translating ribosomes at the ER membrane. It is unclear whether the binding of BiP and ribosomes to ERdj1 occur simultaneously or sequentially. Dudek et al. showed that ERdj1 can simultaneously interact with the ribosome and BiP and that the binding of BiP promotes the re-initiation of translation
[4]. On the other hand, experimental data from surface plasmon resonance show that the binding of BiP to ERdj1 prevents the binding of ERdj1 to ribosomes
[5], indicating that the ERdj1/ribosome complex needs to be established before BiP can bind to it. In any case, BiP abolishes the inhibitory effect of ERdj1 on protein translation, whether or not the binding of BiP to ERdj1 occurs simultaneously or sequentially
[4][5]. It is also still unclear how BiP binding to ERdj1 is signaled to the cytosol to release the translational arrest
[3]. Additionally, the data from surface plasmon resonance, which demonstrated that the prebinding of BiP to ERdj1 inhibits ribosome binding to ERdj1, are difficult to understand, given the fact that ERdj1 has a much higher affinity for ribosomes (K
D 30 pM) than for BiP (K
D 0.57µM)
[1][7]. A possible mechanism could be that the prebinding of BiP to ERdj1 alters the affinity of ERdj1 for ribosomes. The fact that ERdj1 can inhibit the translation until BiP binds to ERdj1 could present a mechanism by which the ER luminal balance of the chaperone and protein levels is ensured. When a large number of unfolded proteins prevails within the ER, BiP is released from ERdj1 to bind to client proteins, thereby inducing an ERdj1-mediated translational arrest (
Figure 1a). When client proteins are folded successfully, BiP is again available to bind to ERdj1 so that the translation can progress
[4] (
Figure 1b). Translational arrest is a known mechanism of reducing ER stress during the UPR, mainly mediated by the ER stress sensor PERK
[8] (
Figure 1). The PERK-dependent phosphorylation of eIF2α inhibits the formation of the translational initiation complex
[8][9]. This results in the inhibition of general, eIF2α-dependent protein translations, so that the protein load in the ER is reduced
[8]. Given this analogy, ERdj1 presumably acts as an additional sensor of ER stress
[4]. The dissociation constant of the ERdj1/BiP complex in the presence of ATP is 0.57 µM, whereas the dissociation constant of the PERK luminal domain/BiP complex is 0.83 µM
[5][10], which allows a fine tuning of translational arrest to the amount of accumulating, BiP-consuming unfolded proteins. According to these results, BiP would dissociate more easily from PERK than from ERdj1, and ERdj1-mediated translational inhibition would sequentially follow PERK-mediated translational inhibition (
Figure 1b). Yet, it has to be noted that the experimental conditions do not really reflect the in vivo situation. Neither of the experiments were performed with PERK or ERdj1 in their original topology as integral membrane proteins
[5][10]. Additionally, different methods were used to determine the affinity of BiP to ERdj1 or to PERK using surface plasmon resonance
[5] or microscale thermophoresis
[10], respectively.
In conclusion, determination of the dissociation constants of PERK/BiP and ERdj1/BiP in their original topologies with identical experimental methods should be performed, since these values are critically important for a reliable evaluation of the discussed sequential BiP release from its stress sensors ERdj1 and PERK.
The co-chaperone ERdj2, in conjunction with Sec62, also controls protein translation (
Figure 1). Like ERdj1, Sec62 associates with emerging proteins close to the tunnel exit of the 60S ribosomal subunit
[3][7]. Due to its association with the translocon-associated protein Sec62, ERdj2 is also termed Sec63
[7][11][12]. The interaction of Sec62 and ERdj2 has been investigated by pulldown assays and surface plasmon resonance spectroscopy. Accordingly, the C-terminal residues (aa734–aa760) of ERdj2 interact with the N-terminal residues (aa11–aa155) of human Sec62. The weak binding of both proteins was shown to be strengthened by CK2-mediated phosphorylation
[7][11][12][13]. Pulldown experiments revealed that the binding of Sec62 to ribosomes weakens the binding of Sec62 to ERdj2 and that Sec62 is displaced from ERdj2 upon binding to ribosomes
[7]. This is in accordance with the data showing a higher affinity of Sec62 to ribosomes (K
D 0.13 nM) than to ERdj2 (K
D 5 nM)
[7][14]. Furthermore, the ribosome-binding site is close to the N-terminal, ERdj2-binding region of Sec62, as was shown in in vitro ribosome binding assays and fractionation experiments
[7]. Interestingly, co-immunoprecipitation experiments using microsomal extracts could precipitate Sec62 due to its binding to ERdj2 but not vice versa
[11][12]. The reason for this is unknown. Yet, the experiments in which the co-immunoprecipitation of ERdj2 with Sec62 failed were performed with antibodies, which were directed against the C-terminus of Sec62
[11][12]. It can be assumed that the binding of the precipitating antibody induces a conformational change at the N-terminus of Sec62, thereby disabling the binding of Sec62 to ERdj2
[7].
Taken together, two functional pools of Sec62 seem to exist at the ER membrane, with Sec62 being either associated with or displaced from ERdj2. When displaced from ERdj2, Sec62 is bound to ribosomes, a conformation in which protein synthesis and translocation is inhibited.
Interestingly, a functional redundancy exists between ERdj1 and Sec62 in that both proteins bind to the same region at the ribosomal tunnel exit. The preincubation of ribosomes with the C-terminus of ERdj1 (ERdj1C) prevents binding of the N-terminus of Sec62 (Sec62N) to ribosomes
[7]. The binding of Sec62N to a translating ribosome can inhibit the initiation of protein translation in a strikingly similar manner as the binding of ERdj1 to translating ribosomes. The interaction of Sec62N with ribosomes is, as the interaction of ERdj1 with ribosomes, RNase- and salt-sensitive and can be abolished at a KCl concentration of 300 mM
[5]. Due to the functional similarity between ERdj1 and the Sec62N–ERdj2 complex, overlapping functions at the ER membrane might explain the fact that the loss of ERdj2 function does not cause a lethal phenotype
[5]. In this context, it would be interesting to examine the effect of ERdj1/Sec62 or ERdj1/ERdj2 double knockouts. Furthermore, it would be interesting to know how ERdj2 binding to Sec62 affects Sec62 binding to ribosomes and how BiP binding to ERdj2 affects the ERdj2/Sec62/ribosome complex.
2. ERdj6 Reinitiates Translation at the End of a Stress Period
Apart from ERdj1 and ERdj2, additional translational control is exerted by the co-chaperone ERdj6, which is upregulated in ER-stressed cells
[15][16] (
Figure 1). The upregulation of ERdj6 was reported to inhibit the amount of phosphorylated PERK, as well as phosphorylated eIF2α, in cells, thus alleviating the inhibitory effect of PERK on RNA translation and protein synthesis
[15]. The stimulation of protein synthesis has been shown in HeLa cells, as well as in HEK293 cells, in which the downregulation of ERdj6 results in the downregulation of general protein synthesis
[16][17]. This is interesting, since
ERdj6 transcription and ERdj6 synthesis themselves are upregulated in stressed cells
[15][16]. Accordingly, in stressed cells, the upregulated ERdj6 levels silence PERK, thereby releasing PERK-mediated translational arrest
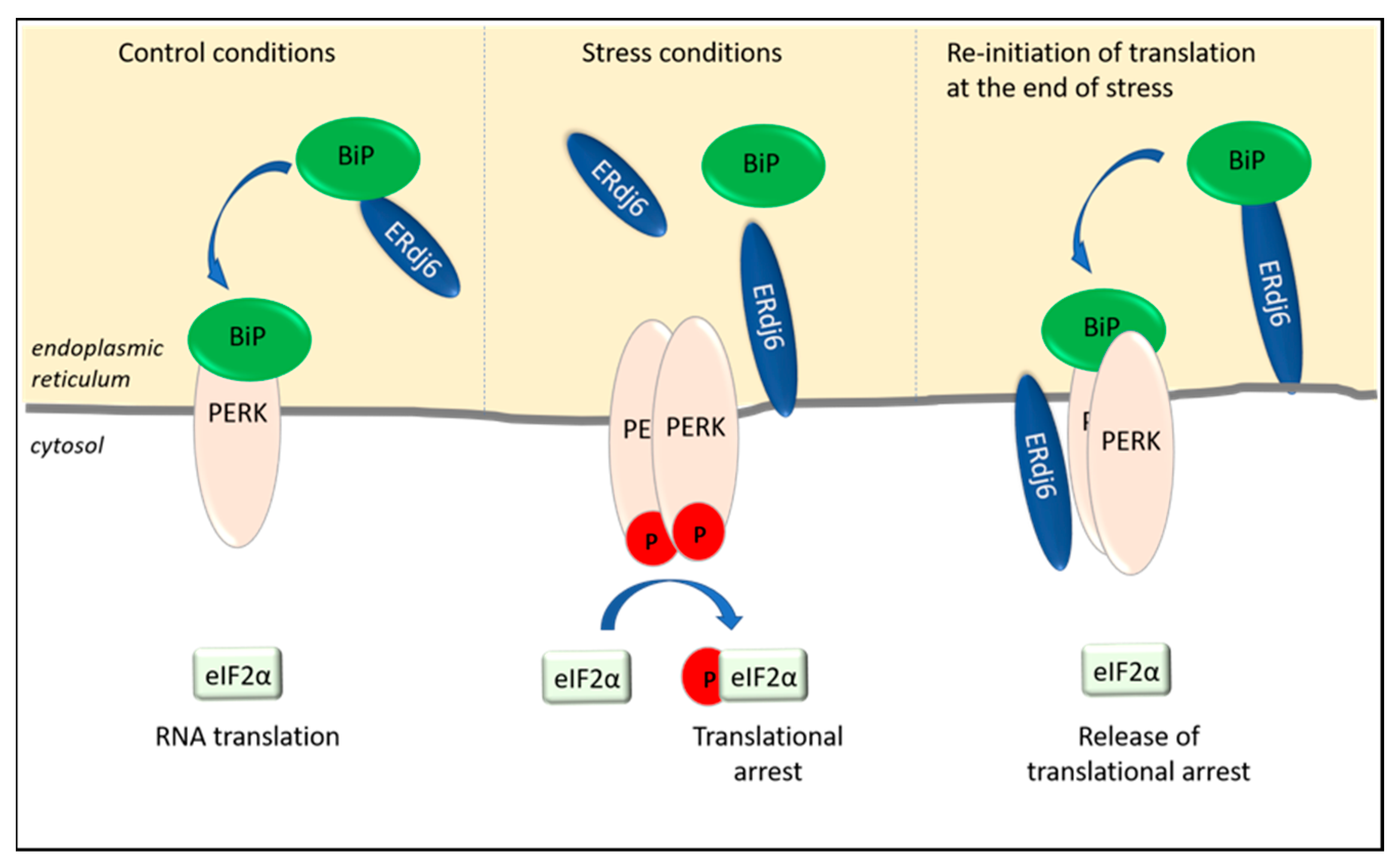
[15] and re-adjusting the cellular metabolism towards increased rates of protein synthesis (
Figure 2). This scenario must be considered as a feedback mechanism to normalize cellular physiology at the end of a stress period. This hypothesis is supported by data indicating a switch in the topology of ERdj6 upon ER stress. In cell lysates of control cells transfected with ERdj6, which was engineered to contain a C-terminal glycosylation site (-CHO mutant), EndoH treatment, as well as a proteinase K experiment, revealed that the entire ERdj6-CHO protein pool—with a cleaved signal sequence—seems to be located in the ER lumen
[17][18]. After 8 h of thapsigargin-mediated ER stress, a membrane-anchored ER luminal pool appears beside the free luminal pool, and after 24 h of thapsigargin treatment, the only protein pool present contains the uncleaved membrane signal, with one-half being glycosylated and the other half unglycosylated and susceptible to proteinase K treatment
[17]. So, while under control conditions, ERdj6 is mainly present as a luminal ER resident protein, the treatment with thapsigargin increases the pool of ER signal peptide-uncleaved ERdj6 protein, located luminally as well as cytosolically
[2][17]. Since ERdj6 co-immunoprecipitates with full-length PERK but not with a construct lacking the cytosolic domain of PERK, it was suggested that ERdj6 exerts its inhibitory function on PERK by directly binding to the cytosolic kinase domain of PERK
[15] (
Figure 2).
Figure 2. ERdj6 surveys the activation state of PERK. ERdj6 can adopt various subcellular localizations. Under control conditions, ERdj6 is located luminally and transfers BiP to PERK to keep PERK in a silenced state and to enable eIF2α-dependent RNA translation. In ER-stressed cells (as observed after 8 h thapsigargin treatment), ERdj6 is inserted in the ER membrane. Half of the membrane-anchored pool faces the lumen, and half of the pool faces the cytosol. Due to the increased demand of chaperoning activity, BiP dissociates from the luminal domain of PERK and enables the activation and autophosphorylation of PERK. The subsequent phosphorylation of eIF2α inhibits eIF2α-dependent translational processes. The translational arrest can be released by interaction of the cytosolic ERdj6 pool with the C-terminus of PERK (see text).
On the other hand, the luminal ERdj6 pool also has the potential to silence PERK by promoting the formation of the BiP/PERK complex. According to the estimated dissociation constants, BiP has a greater affinity to PERK (K
D 0.83 µM) than to ERdj6 (K
D 5 µM)
[10][19]. Hence, as schematically shown in
Figure 2, ERdj6 could recruit and transfer BiP to PERK similar to the models suggested for ERdj2 (see above), as well as for ERdj4, which was shown to act as the BiP donor for IRE1, thereby keeping the respective signaling pathways in a silenced state
[20][21]. Still, no experimental evidence has been presented so far that would support such a model.
All in all, the Hsp40 co-chaperones ERdj1 and ERdj2 (as the ERdj2/Sec62 complex) arrest protein translation in the absence of available BiP by direct association with the ribosome, while elevated ERdj6 levels in stressed cells promote protein translation by alleviating PERK-mediated translational arrest.
 +1 credit
+1 credit