Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hamza Yazdani | -- | 5444 | 2022-07-27 16:10:47 | | | |
| 2 | Hamza Yazdani | -3876 word(s) | 1568 | 2022-07-28 05:41:11 | | | | |
| 3 | Vivi Li | Meta information modification | 1568 | 2022-07-28 08:40:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kaltenmeier, C.; Wang, R.; Popp, B.; Geller, D.; Tohme, S.; Yazdani, H.O. Immuno-Inflammatory Signals in Liver Ischemia-Reperfusion Injury. Encyclopedia. Available online: https://encyclopedia.pub/entry/25588 (accessed on 09 August 2026).
Kaltenmeier C, Wang R, Popp B, Geller D, Tohme S, Yazdani HO. Immuno-Inflammatory Signals in Liver Ischemia-Reperfusion Injury. Encyclopedia. Available at: https://encyclopedia.pub/entry/25588. Accessed August 09, 2026.
Kaltenmeier, Christof, Ronghua Wang, Brandon Popp, David Geller, Samer Tohme, Hamza O. Yazdani. "Immuno-Inflammatory Signals in Liver Ischemia-Reperfusion Injury" Encyclopedia, https://encyclopedia.pub/entry/25588 (accessed August 09, 2026).
Kaltenmeier, C., Wang, R., Popp, B., Geller, D., Tohme, S., & Yazdani, H.O. (2022, July 27). Immuno-Inflammatory Signals in Liver Ischemia-Reperfusion Injury. In Encyclopedia. https://encyclopedia.pub/entry/25588
Kaltenmeier, Christof, et al. "Immuno-Inflammatory Signals in Liver Ischemia-Reperfusion Injury." Encyclopedia. Web. 27 July, 2022.
Copy Citation
Ischemia reperfusion injury (IRI) is a major obstacle in liver resection and liver transplantation. The initial step of IRI is mediated through ischemia which promotes the production of reactive oxygen species in hepatic cells. This promotes the activation of pro-inflammatory signaling cascades and the production of damage-associated molecular patterns (DAMPs). During the ischemic phase, DAMPs are released into the circulation by stressed cells upon reperfusion that further attracts neutrophils and other immune cells to the site of tissue injury.
ischemia-reperfusion injury
ROS
Kupffer cells
neutrophils
NETs
DAMPs
platelets
thrombosis
miRNA
1. Introduction
Liver ischemia and reperfusion injury is a frequent consequence in a variety of surgical procedures, and it is well established that IRI is a major cause of morbidity and mortality in liver resection and transplantation surgery [1][2][3]. Prolonged IRI reduces tissue oxygenation and subsequent transition to anaerobic metabolism in hepatic resident cells. During transplantation, prolonged ischemic time is associated with delayed graft function, early rejection and increased risk of complications [4]. The pathogenesis of IRI is fueled by several different mechanisms such as oxidative stress, inflammatory responses, Kupffer cell activation, and neutrophil migration and activation.
It is known that ongoing ischemia promotes the build-up of waste products as several cellular functions fail to maintain homeostasis under oxygen deprivation. Stressed cells start producing reactive oxygen species (ROS) that increase DNA and organelle damage. Prolonged ischemia increases the production of damage-associated molecular patterns (DAMPs) that are released into the surrounding tissue upon cell death [5]. These DAMPs can increase pro-inflammatory signaling cascades in surrounding cells as well as activate the complement system (Figure 1). With restoration of blood flow, several pathways are initiated that further promote cellular damage. DAMPs are flushed into the circulation and thereby promote the activation and recruitment of immune cells to the site of tissue injury [6][7]. Following reperfusion, neutrophils are among the first cells to enter the liver and there is overwhelming evidence that excessive neutrophil infiltration contributes to the pathogenesis of IRI.
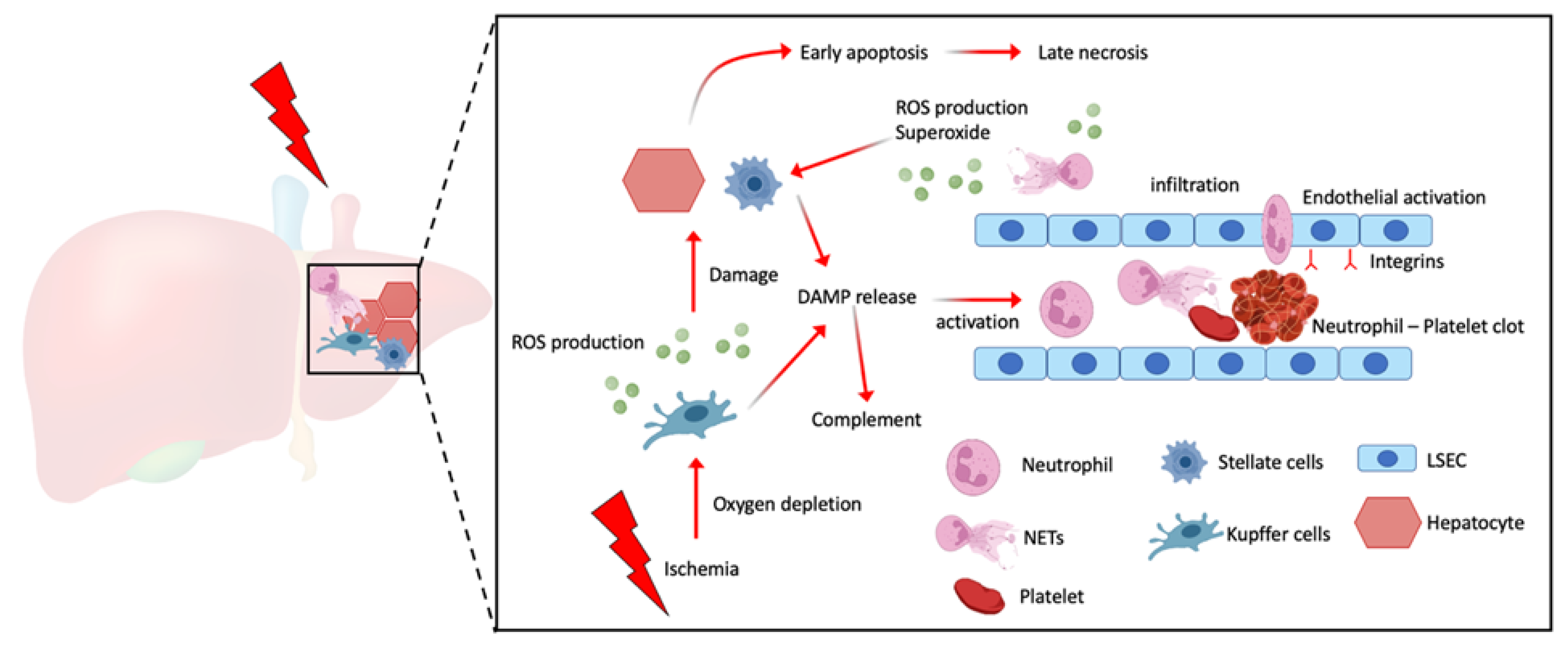
Figure 1. Schematic illustration showing prolonged ischemia reduces hepatic tissue oxygenation resulting in increased ROS production and DAMP release by both Kupffer cells and hepatic stellate cells which can induce hepatic cell death. These DAMPs can play a role as an innate immune cell attractant and activator, especially neutrophils to undergo NETosis. NETs can further promote platelet activation and capture resulting in microthrombi.
2. Reactive Oxygen Production and Its Regulation during Ischemia
During the initial stage of IR, Kupffer cells are the main producers of reactive oxygen species (ROS) and other pro-inflammatory mediators including tumor necrosis factor TNF-α and interleukin (IL)-6 [8]. ROS production can promote lipid peroxidation and increase cell membrane and mitochondrial electron leakage. Furthermore, ROS increases signal transduction pathways, caspase activation and promotes cell death of hepatic stellate, Kupffer and endothelial cells [9].
Reperfusion leads to a dramatic rise in oxygen delivery which exceeds the rate at which cellular metabolism is capable of working, thereby producing free radicals that promote DNA and cellular damage. In fact, the main driver of ROS production during IRI is the massive accumulation of succinate within the mitochondria. Upon reperfusion, succinate is oxidized within the electron transport chain and ROS is produced [10].
One way to regulate ROS production is through the phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) signaling pathway. The PI3K/AKT pathway is involved in various cellular physiological and pathological functions. This pathway is activated during the early phase of IRI where it plays a role in anti-apoptosis, -inflammation and -oxidation. PI3K is a well-studied redox sensitive kinase that is activated in an acidic environment where increased levels of ROS can be found. The activated PI3K/Akt signaling pathway promotes the generation of heme-oxygenase-1 (HO-1) by upregulating the expression of Nrf2 which decreases the oxidative stress following IRI [8][9].
3. Cell Death Signaling during IRI
During the ischemic phase of IRI, the two predominant cell populations that are injured are hepatocytes and sinusoidal endothelial cells (SECs). Apoptosis and necrosis are both seen during IRI depending on the duration of ischemia and time of reperfusion. The hallmarks of apoptosis include shrinkage of the cell, chromatin condensation, nuclear fragmentation and the formation of apoptotic bodies. Necrosis can be characterized by the presence of mitochondrial and cell swelling with a loss of plasma membrane integrity and leakage [11]. Interestingly, the intensity of a given stimulus can have varying effects and can promote the activation of apoptotic or necrotic pathways. Several studies have shown the presence of apoptotic hepatocytes following IRI by using TUNEL staining. However, these studies have also shown that despite the presence of apoptotic pathways in hepatocytes, the final form of cell death following prolonged ischemia and reperfusion remains necrosis [12][13]. Apoptosis is an extremely complex pathway with a close relation to free radical production, intracellular calcium overload, cytokine activation, caspase and B lymphocyte tumor-2 (Bcl-2) gene expression. When cells receive apoptosis signal stimulations, the Bcl-2 protein regulates the permeability of the mitochondrial membrane. Apoptosis leads to irreversible opening of the mitochondrial permeability transition pore (mPTP), which leads to the release of cytochrome C into the cytoplasm. Within the cytoplasm, cytochrome C interacts with apoptosis protein activated factor 1 (Apaf-1) which activates the caspase 9 precursor though an auto-cleavage process. This complex together with caspase 3 can further induce apoptotic cell death [14][15].
4. Cell Signaling in Recruiting Neutrophils during IRI
Under hypoxic stress, Kupffer cells increase the production of DAMPs such as high-mobility group B1 (HMGB1), S100, heat shock proteins and circulating DNA/RNA that can all trigger a variety of inflammatory immune responses. HMGB1 can activate Toll-like receptor 4 (TLR4) which in turn triggers the recruitment of MyD88 promoting several downstream signaling pathways leading ultimately to the production of NF-κB, TNF-α and IL-6 which promote the upregulation of adhesion molecules on the luminal site of endothelial cells to aid with migration of neutrophils. Studies have shown that PI3K can inhibit the activation of the forementioned pathway and thereby alleviate IRI [16][17]. Shen et al. have shown that the activation of the PI3K/Akt signaling pathway lead to increased expression of anti-inflammatory cytokines IL-4 and IL-10 and decreased expression of TNF-α, IL-6 and IL-1β to ultimately reduce the IRI-induced liver inflammatory response [18].
Following the reperfusion of blood into the previously ischemic tissues, neutrophils are one of the first cell types that are activated via circulating DAMPs such as HMGB1 and ATP. As neutrophils are called to the area of inflammation, they adhere to the endothelial wall and transmigrate into the damaged tissue. Weibel-Palade bodies, which are produced by endothelial cells, secrete P-selectin, an important chemokine in neutrophil trafficking. P-selectin increases endothelial cell permeability which in turn slows down neutrophils, as they travel through the bloodstream, and allows them to roll along the endothelial cell wall [5]. This slowed neutrophil transit allows intercellular adhesion molecule (ICAM)-1 to promote adhesion of the neutrophil to the endothelial cell wall. ICAM-1 binds to integrins, which are expressed on the cell surface of activated neutrophils and causes neutrophils to adhere tightly to the blood vessel wall. From there, neutrophils transmigrate across the endothelium and travel towards chemokines produced by damaged tissues.
5. Neutrophil Extracellular Traps in regulating IRI
As previously mentioned, neutrophils play an important role in initiation and maintenance of liver IRI. Neutrophils maintain the pro-inflammatory state by production of proteolytic enzymes, arachidonic acid metabolites and ROS. Several metabolites and DAMPs can promote the activation and formation of neutrophil extracellular traps (NETs) in neutrophils. Activated neutrophils release MPO and other proteases that facilitate the formation of superoxide causing direct damage of liver endothelial cells. In addition, NETs which are DNA-linked, web-like structures can further facilitate damage by crosslinking with platelets to increase thrombosis, activation of immune cells or the complement system [19][20].
NETs have also been seen to play a role in clotting and thrombus formation. In a murine model of liver IRI, researchers' laboratory has shown that induction of NETs can further activate platelets results in systemic immune-thrombosis and distant organ injury [21][22]. The study showed that 1 h of ischemia followed by 6 h of reperfusion lead to activation of platelets and increased neutrophil platelet aggregation. These aggregates formed micro-thrombi within the liver following IRI. When NETs were blocked using DNAse-1 therapy, immunological thrombi and organ damage were significantly reduced. Interestingly, when utilizing platelet-specific TLR4 KO animals, researchers found significant decreased organ damage, with lower circulating platelet activation and platelet-neutrophil aggregates following liver I/R. This effect of TLR4-dependent platelet activation is thought to be mediated through HMGB1 released by neutrophils [22].
Fuchs et al. have furthermore demonstrated the role of DNA as well as histones H3 and H4 in NET-induced platelet aggregation in vitro [23]. However, in their study, they found that NET-dependent platelet aggregation was shown to be unaffected by DNAse or heparin treatment, which would suggest a mechanism independent of the DNA structure and thrombin. They furthermore showed that blocking of cathepsin G, which is contained within the NET chromatin, led to reduced expression of CD62 and phosphatidylserine and decreased aggregation. This finding was further supported by Nemmar et al. who showed that neutrophil-specific elastase increased the cathepsin G-induced platelet aggregation mediated by neutrophils [24]. In addition, PMA-treated neutrophils have been shown to release microparticles that can attach themselves to NETs via phosphatidylserine residues Figure 2.
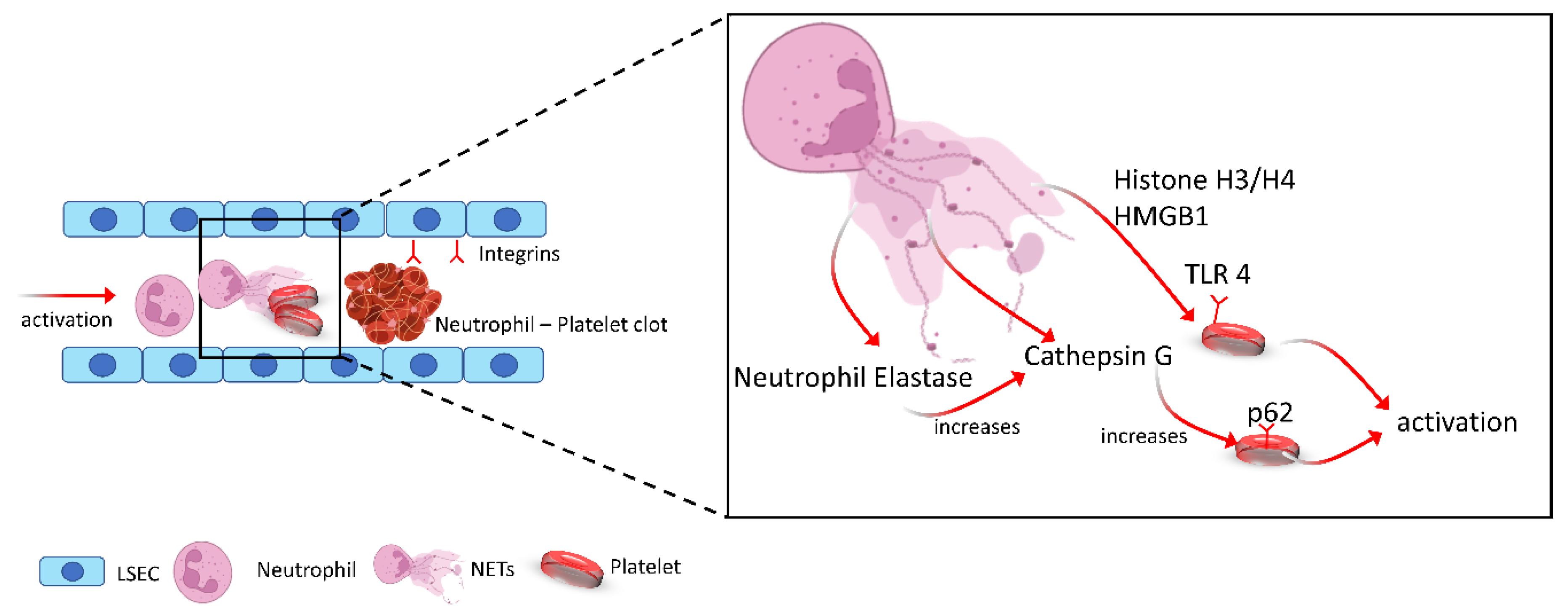
Figure 2. Schematic illustration showing increase in the release of NET-decorated proteins such as neutrophil elastase, cathepsin G, HMGB1 and histone upon NETosis. These proteins induce platelet activation through TLR4 and p62 pathways, resulting in the formation of microthrombi within the liver and promote IR induce liver injury.
References
- Nakamura, K.; Kageyama, S.; Kupiec-Weglinski, J.W. The Evolving Role of Neutrophils in Liver Transplant Ischemia-Reperfusion Injury. Curr. Transplant. Rep. 2019, 6, 78–89.
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, A.K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380.
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for cancer: A trigger for metastases. Cancer Res. 2017, 77, 1548–1552.
- Hudcova, J.; Scopa, C.; Rashid, J.; Waqas, A.; Ruthazer, R.; Schumann, R. Effect of early allograft dysfunction on outcomes following liver transplantation. Clin. Transplant. 2017, 31, e12887.
- De Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62.
- Hu, Q.; Wood, C.R.; Cimen, S.; Venkatachalam, A.B.; Alwayn, I.P.J. Mitochondrial Damage-Associated Molecular Patterns (MTDs) are Released during Hepatic Ischemia Reperfusion and Induce Inflammatory Responses. PLoS ONE 2015, 10, e0140105.
- Hirao, H.; Dery, K.J.; Kageyama, S.; Nakamura, K.; Kupiec-Weglinski, J.W. Heme Oxygenase-1 in liver transplant ischemia-reperfusion injury: From bench-to-bedside. Free Radic. Biol. Med. 2020, 157, 75–82.
- Wang, M.; Zhang, J.; Gong, N. Role of the PI3K/Akt signaling pathway in liver ischemia reperfusion injury: A narrative review. Ann. Palliat. Med. 2022, 11, 806–817.
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A paradoxical relationship. Trends Cell Mol. Biol. 2012, 7, 1–10.
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435.
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537.
- Gujral, J.S.; Bucci, T.J.; Farhood, A.; Jaeschke, H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: Apoptosis or necrosis? Hepatology 2001, 33, 397–405.
- Yang, M.; Antoine, D.J.; Weemhoff, J.L.; Jenkins, R.E.; Farhood, A.; Park, B.K.; Jaeschke, H. Biomarkers distinguish apoptotic and necrotic cell death during hepatic ischemia/reperfusion injury in mice. Liver Transplant. 2014, 20, 1372–1382.
- Martinou, J.-C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92–101.
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686.
- Guha, M.; Mackman, N. The Phosphatidylinositol 3-Kinase-Akt Pathway Limits Lipopolysaccharide Activation of Signaling Pathways and Expression of Inflammatory Mediators in Human Monocytic Cells. J. Biol. Chem. 2002, 277, 32124–32132.
- Li, X.; Wu, Y.; Zhang, W.; Gong, J.; Cheng, Y. Pre-conditioning with tanshinone IIA attenuates the ischemia/reperfusion injury caused by liver grafts via regulation of HMGB1 in rat Kupffer cells. Biomed. Pharmacother. 2017, 89, 1392–1400.
- Shen, Y.; Shen, X.; Cheng, Y.; Liu, Y. Myricitrin pretreatment ameliorates mouse liver ischemia reperfusion injury. Int. Immunopharmacol. 2020, 89, 107005.
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536.
- Tan, C.; Aziz, M.; Wang, P. The vitals of NETs. J. Leukoc. Biol. 2021, 110, 797–808.
- Dyer, M.R.; Chen, Q.; Haldeman, S.; Yazdani, H.; Hoffman, R.; Loughran, P.; Tsung, A.; Zuckerbraun, B.S.; Simmons, R.L.; Neal, M.D. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci. Rep. 2018, 8, 2068.
- Zhang, H.; Goswami, J.; Varley, P.; Van Der Windt, D.J.; Ren, J.; Loughran, P.; Yazdani, H.; Neal, M.D.; Simmons, R.L.; Zhang, J.; et al. Hepatic surgical stress promotes systemic immunothrombosis that results in distant organ injury. Front. Immunol. 2020, 11, 987.
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85.
- Nemmar, A.; Hoylaerts, M.F. Neutrophil Cathepsin G Enhances Thrombogenicity of Mildly Injured Arteries via ADP-Mediated Platelet Sensitization. Int. J. Mol. Sci. 2022, 23, 744.
More
Information
Subjects:
Surgery
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
3 times
(View History)
Update Date:
28 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No