Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pameila Paerhati | -- | 2200 | 2022-07-27 06:01:42 | | | |
| 2 | Sirius Huang | Meta information modification | 2200 | 2022-07-27 07:36:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Paerhati, P.; Liu, J.; Jin, Z.; Jakoš, T.; Zhu, S.; Qian, L.; Zhu, J.; Yuan, Y. ATF5 Function in Regulating Cell Stress and Survival. Encyclopedia. Available online: https://encyclopedia.pub/entry/25562 (accessed on 25 July 2026).
Paerhati P, Liu J, Jin Z, Jakoš T, Zhu S, Qian L, et al. ATF5 Function in Regulating Cell Stress and Survival. Encyclopedia. Available at: https://encyclopedia.pub/entry/25562. Accessed July 25, 2026.
Paerhati, Pameila, Jing Liu, Zhedong Jin, Tanja Jakoš, Shunyin Zhu, Lan Qian, Jianwei Zhu, Yunsheng Yuan. "ATF5 Function in Regulating Cell Stress and Survival" Encyclopedia, https://encyclopedia.pub/entry/25562 (accessed July 25, 2026).
Paerhati, P., Liu, J., Jin, Z., Jakoš, T., Zhu, S., Qian, L., Zhu, J., & Yuan, Y. (2022, July 27). ATF5 Function in Regulating Cell Stress and Survival. In Encyclopedia. https://encyclopedia.pub/entry/25562
Paerhati, Pameila, et al. "ATF5 Function in Regulating Cell Stress and Survival." Encyclopedia. Web. 27 July, 2022.
Copy Citation
Activating transcription factor 5 (ATF5) belongs to the activating transcription factor/cyclic adenosine monophosphate (cAMP) response element-binding protein family of basic region leucine zipper transcription factors. ATF5 plays an important role in cell stress regulation and is involved in cell differentiation and survival, as well as centrosome maintenance and development.
ATF5
cell stress
mitochondrial UPR
cell survival
1. Biological ATF5 Functions
1.1. Cell Differentiation and Tissue Development
Activating transcription factor 5 (ATF5) regulates stem cell differentiation and is essential for tissue development. In the development of cerebral neurons, ATF5 inhibits the differentiation and maintains the proliferative state of neural progenitor cells into neurons, astrocytes, and oligodendroglia [1][2][3]. ATF5 also regulates cerebellar granule neuron development by increasing the sonic hedgehog protein-mediated proliferation and neuronal differentiation [4]. Several studies on ATF5 knockout mice revealed that ATF5 deficiency leads to neonatal death, olfactory defects, olfactory bulb hypogenesis, behavioral abnormalities, and the abnormal development of the cerebral cortex [5][6][7][8]. In addition to neural development, ATF5 is involved in the regulation of non-neural differentiation. ATF5 mediates the promotion of adipocyte differentiation, hepatocyte differentiation, and the inhibition of osteogenic differentiation, [9][10][11]. A recent study revealed that ATF5 is enriched in odontoblast-related nucleosome-free regions and promotes odontoblast terminal differentiation by increasing the enhancer activity of dentin matrix protein 1 (DMP1), which is mainly expressed in the odontoblast layer and important for bone development [12]. Another study demonstrated that the knockdown of ATF5 in murine erythroid progenitor cells significantly reduced the proliferation of fetal liver erythroid progenitors and inhibited erythroid differentiation [13].
1.2. Centrosome Maturation
ATF5 acts as a structural protein that regulates and maintains centrosome integrity. During the centrosome duplication cycle, ATF5 regulates the pericentriolar material-dependent centriole formation by interacting with pericentrin and polyglutamylated tubulin [14]. ATF5 SUMOylation occurs in a cell-cycle-dependent manner, and the highest and lowest levels of SUMOylation take place at the G1 and M phases, respectively. The SUMOylation of ATF5 interferes with its association with centrosome proteins at the molecular level. The mutation of SUMOylation sites in ATF5 can improve its accumulation on the centrosomes at the G1 phase and block its dissociation of centrosomes [15]. The centrosome cycle changes usually result in centrosome abnormalities that induce genome instability and tumorigenesis. This mechanism can partially explain ATF5’s regulation of centrosome maturation and its high expression in many types of tumor cells.
1.3. Responses to Cellular Stress
ATF5 participates in the cellular stress response to ER stress, mitochondria dysfunction, arsenite exposure, and proteasome inhibition [16][17]. Abundant evidence exists proving that ATF5 is involved in pro-survival pathways activated under cellular stress, such as the ER unfolded protein response (UPR), mitochondrial UPR (UPRmt), and heat-shock response (HSR). ATF5 over-expression can protect neural, retinal ganglion, chondrocyte, and pancreatic β cells from apoptosis under ER stress [18][19][20][21]. ATF5 also promotes cardiomyocyte survival by activating heat-shock protein 27 (HSP27) under hyperthermia-induced heat-shock stress in cardiomyocytes [22]. Meanwhile, up-regulated levels of ATF5 in mitochondrial dysfunction promote mitochondrial oxidative phosphorylation (OXPHOS) recovery and induce UPRmt by mediating the transcription of mitochondrial chaperones (heat-shock protein 60 (HSP60) and mitochondrial heat-shock protein 70 (mtHSP70)) and proteases (mitochondrial Lon peptidase 1 (LONP1)) to promote survival and recovery through maintaining protein homeostasis under mitochondrial stress [23][24]. Among these, HSP60 and mtHSP70 conjointly promote the correct folding of denatured and newly synthesized polypeptides. LONP1 enhances the degradation of irreversibly damaged proteins by cleaving them and mediates the remodeling of OXPHOS complexes, which are needed for the survival and proliferation of tumor cells [25][26][27].
The protective role of ATF5 in cardiac mitochondrial dysfunction has become clearer with increasing research on the function of ATF5-regulated UPRmt. ATF5 activates UPRmt-induced protection in cardiac ischemia–reperfusion injury (I/R), and the global knockdown of ATF5 results in the failure of the cardioprotective effects of the UPRmt inducers oligomycin or doxycycline in I/R [28]. ATF5 also participates in the activation of the UPRmt-mediated cardioprotective role of tetrahydrocurcumin (THC), a traditional treatment for cardiovascular disease, and in pathological cardiac hypertrophy by forming a signaling axis with peroxisome proliferator-activated receptor-gamma co-activator-1 α (PGC-1α), which is a regulator of mitochondrial quality control and energy metabolism [29]. The aberrant expression of ATF5 has emerged as one of the biomarkers for increased UPRmt activity and improved mitochondrial function. For instance, the significant up-regulation of ATF5 mRNA and protein levels might reflect the UPRmt-enhanced mitochondrial resistance to early cocaine-induced cardiotoxicity. Additionally, a decreased ATF5 expression indicates UPRmt inactivation and insufficient mitochondrial recovery in heart failure with a preserved ejection fraction (HFpEF) [30][31].
2. Transcriptional Regulation and Down-Stream Targets of ATF5
ATF5 expression is regulated at both the transcriptional and translational levels in response to different stresses, subsequently affecting the activation of ATF5 down-stream targets and the related signaling pathways that are involved in cell survival. The translational ATF5 expression in response to environmental stress is regulated by eIF2a phosphorylation, which is in turn regulated by the kinases serine/threonine-protein kinase (GCN2), PERK, or PKR [16]. Along with ATF5, eIF2a enhances ATF4 translation, which is necessary for increased ATF5 transcription. In addition, several other up-stream transcriptional factors can activate ATF5 expression, thereby promoting the expression of pro- and anti-survival target genes that regulate the cell survival pathways in cancer development and during cellular stress, inflammation, and metabolic maintenance.
2.1. Survival Pathways in Cancer Development
Accumulating evidence indicates that ATF5 regulates tumor survival and development by mediating a myriad of survival pathways, such as apoptosis, autophagy, tumorigenesis, migration, and invasion (Figure 1). In the anti-apoptotic and anti-autophagy pathways, ATF5 transcription is enhanced by cAMP-responsive element-binding protein 3-like 2 (CREB3L2), whose expression is up-regulated by the RAS/MAPK and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling cascades, which are activated by fibroblast growth factor receptor substrate 2 (FRS2) and p21-activated kinase 1 (PAK1) [32]. ATF5 then activates the transcription of the anti-apoptotic gene myeloid cell leukemia sequence 1 (MCL1) transcription by binding its promoter and increasing the malignant glioma survival through the CREB3L2/ATF5/MCL1 anti-apoptotic pathway. ATF5 can also activate another anti-apoptotic gene, B cell leukemia/lymphoma 2 (BCL2) transcription, therefore, increasing glioblastoma and breast cancer survival. The oncoprotein BCR-ABL activates the PI3K/AKT signaling cascade, which inhibits the ATF5 transcriptional repressor forkhead box O4 (FOXO4) transcription to increase ATF5 expression. Then, the transcription of the anti-autophagy gene mammalian rapamycin target (mTOR) is triggered, resulting in the suppression of autophagy in BCR-ABL-transformed myeloid leukemia cells [33]. Moreover, ATF5 is a down-stream effector of protein arginine methyltransferase 1 (PRMT1), which is an essential regulator of neuroblastoma cell survival [34]. ATF5 is significantly down-regulated in PRMT1-depleted cells, and the over-expression of ATF5 can rescue the neuroblastoma cells from PRMT1 depletion-induced apoptosis.
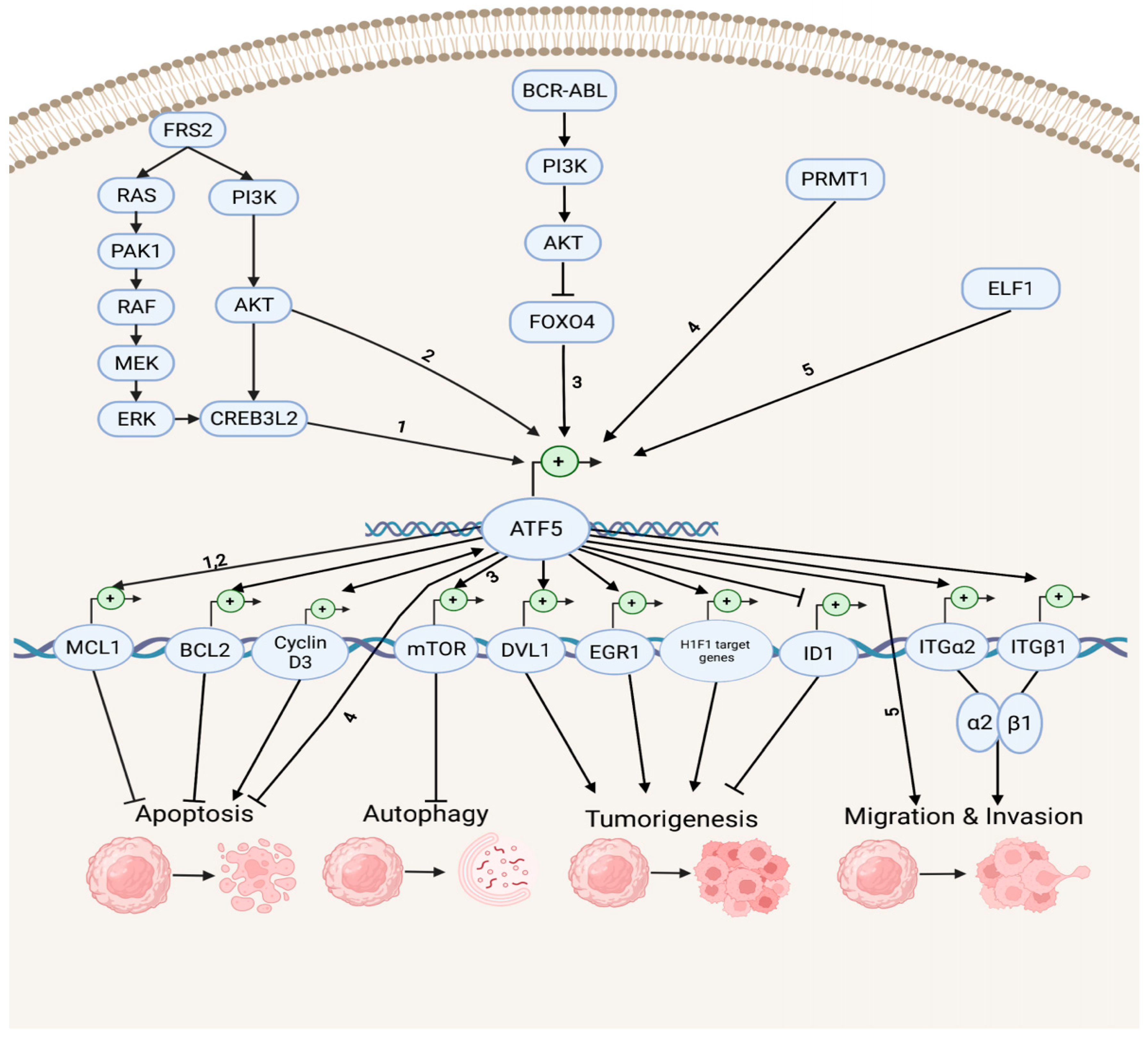
Figure 1. Survival pathways in cancer development. Depicted are up-stream activators of ATF5 and its down-stream target genes that regulate diverse cellular processes, including anti-apoptosis, apoptosis, anti-autophagy, tumorigenesis, migration, and invasion. The numbers represent the known connections between related elements of the signaling pathways.
In the tumorigenesis and migration pathways, ATF5 transcription can be activated by the tumor cell growth and migration promoter E74-like E26 transforming sequence (ETS) transcription factor (ELF1), increasing the malignancy of gliomas [35]. ATF5 directly activates the transcription of the disheveled segment polarity protein gene (DVL1), an essential regulator of the Wnt/β-catenin down-stream effectors (e.g., proto-oncogenes basic helix–loop–helix (bHLH) transcription factor MYC and activating protein (AP-1) transcription factor subunit JUN, cell cycle control gene cyclin D1, as well as non-kinase transmembrane proteoglycan gene CD44), which then mediates the Wnt/β-catenin pathway in bladder cancer and promotes tumorigenesis [36]. ATF5 also increases the hypoxia-inducible factor 1 (HIF1) transcriptional complex activity by binding to HIF1α and promoting the transcription of HIF1 target genes, including 3-phosphoinositide-dependent protein kinase gene (PDK1), carbonic anhydrase (CA9), plasminogen activator inhibitor (PAI1), and vascular endothelial growth factor (VEGFA) genes. This further activates HIF1-mediated tumorigenic signaling and tumor angiogenesis, which can be inhibited by d/nATF5 in vivo esophageal cancer models [37]. Furthermore, ATF5 initiates expression of its pro-invasion down-stream targets, the collagen receptor integrin α2β1 genes ITGα2 and ITGβ1, which can bind to collagen type I, E-cadherin, and matrix metalloproteinase to induce tumor invasion in several human cancer cell lines, such as lung, breast, cervical, gastric, fibrosarcoma, and pancreatic cancers [38][39].
ATF5 can stimulate the proapoptotic pathways in cancer (Figure 1). It can bind to the inhibitor of DNA-binding helix–loop–helix (HLH) protein gene (ID1) promoters at the CRE site and inactivate ID1 transcription, repressing cell proliferation and leading to a G2-M cell cycle arrest during hepatocellular carcinoma (HCC) development [40]. ATF5 can also bind to the cyclin D3 gene promoter and increase its transcription via an interaction with early two-factor family transcription factor 1 (E2F1), resulting in increased cisplatin-induced apoptosis in human cervical cancer cells [41]. ATF5 and cyclin D3 interact in those tumor cells through a positive feedback loop in which cyclin D3 enhances the ATF5 transcription [42].
2.2. Survival Pathways during Cell Stress and Inflammation
ATF5 transcription is also associated with pro- and anti-survival pathways during ER, mitochondrial, and other types of cellular stresses (Figure 2). ATF5 represses ER stress-induced apoptosis through the BBF2H7 (CREB3L2)/ATF5/MCL1 anti-apoptotic pathway in chondrocyte differentiation [21]. Conversely, ATF5 maintains protein homeostasis by co-stimulating BCL2 homology 3 (BH3)-only proapoptotic protein (NOXA)-regulated proapoptotic pathways with ATF4 and DNA damage-inducible transcript 3 (CHOP) [43]. During different stress conditions (e.g., ER unfolded protein, nutrient deprivation, and oxidative damage), the translation of ATF4 and CHOP is enhanced by the phosphorylation of eIF2, and then both bind to the ATF5 promoter and enhance ATF5 transcription. Then, CHOP and ATF5 jointly stimulate the NOXA, thus, resulting in increased cell apoptosis in order to maintain protein homeostasis. ATF5 is also activated by mitochondrial nuclear retrograde regulator 1 (MNRR1), a regulator of mitochondrial metabolism, and induces mitophagy and autophagy to maintain mitochondrial homeostasis during ER stress-induced UPRmt in a MNRR1/ATF5-dependent manner [44].
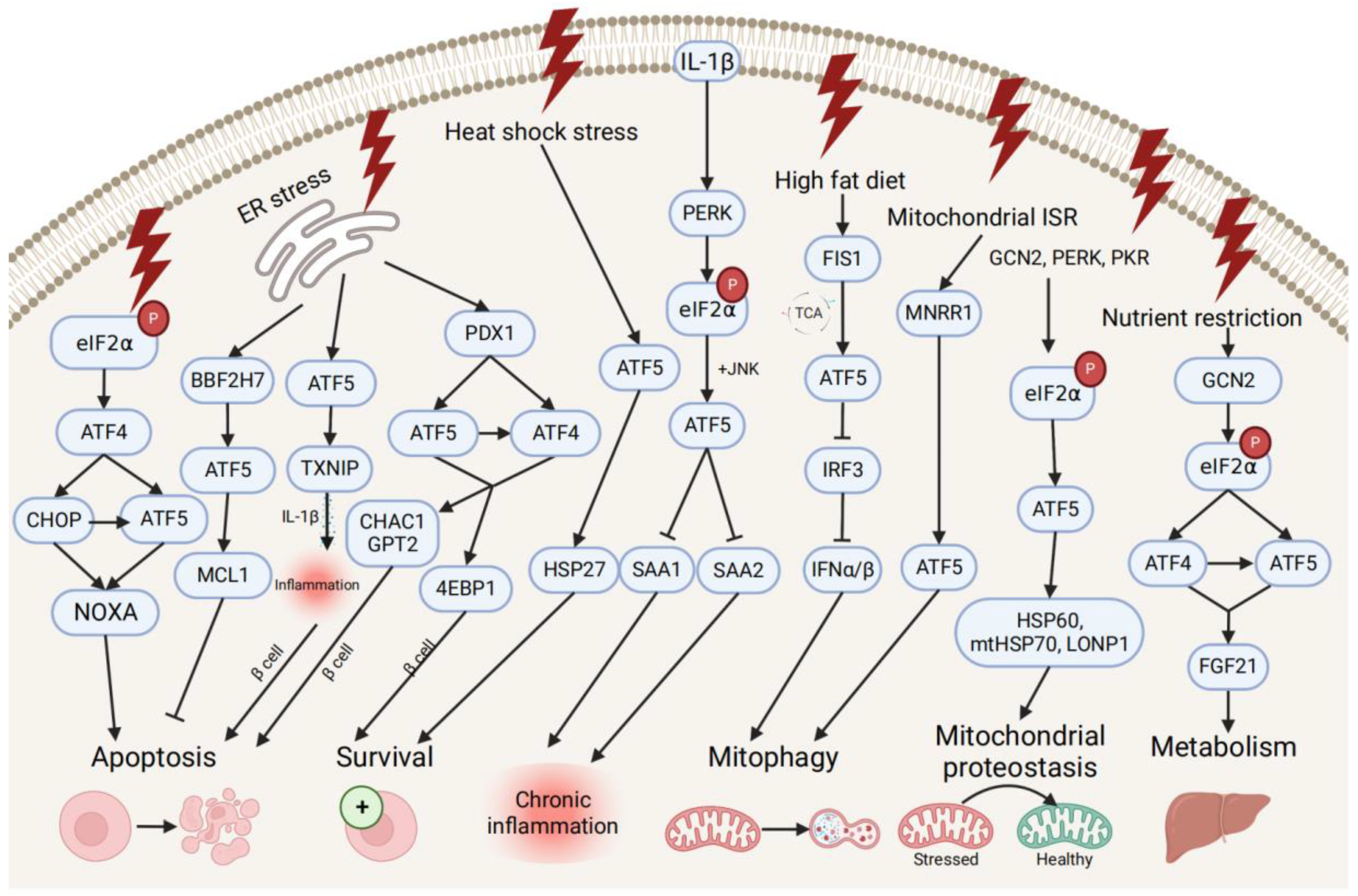
Figure 2. Pro- and anti-survival pathways during cellular stress and inflammation. The up-stream activators of ATF5 are shown, along with target genes that respond to diverse cellular stressors through the induction of anti-apoptotic or apoptotic signals and pathways that promote cell survival, regulate metabolism, chronic inflammation, and mitophagy, and maintain mitochondrial proteostasis.
ATF5 might be involved in the developmental pathway of diabetes by regulating pancreatic β cell survival during cellular stresses and metabolic homeostasis. During ER stress, the human diabetes gene pancreatic and duodenal homeobox 1 (PDX1)-induced ATF4 and ATF5 activation restores the cellular homeostasis and the survival of pancreatic β cells by activating the expression of the protein translation suppressor gene eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) through the PDX1–ATF transcriptional complex [20][45]. On the other hand, ATF5 mediates β cell apoptosis by activating the transcription of the proapoptotic glutathione-degrading enzymes cation transport regulator 1 (CHAC1) and glutamate pyruvate transaminase 2 (GPT2), also through the PDX1–ATF axis, by up-regulating the expression of the thioredoxin interacting protein (TXNIP), which can trigger the inflammasome into producing proinflammatory cytokines (e.g., IL-1β) and inducing inflammation under ER stress [45][46]. Furthermore, ATF5 also participates in insulin resistance and increases lipogenesis through the β-catenin signal activator compound 21 (CP21)-induced Wnt/β-catenin pathway in hepatocytes, as well as influences chronic inflammation and lipid metabolism by down-regulating the expression of serum amyloid genes SAA1 and SAA2, which are overproduced in response to IL-1β-induced inflammatory stress [47][48].
Additionally, ATF5 can promote metabolic maintenance. ATF5’s transcription is stimulated by the mitochondrial fission 1 (FIS1)-triggered accumulation of the tricarboxylic acid cycle (TCA) intermediate under high-fat diet-induced mitochondrial stress in liver cells [49]. The over-expression of ATF5 causes the inhibited transcription of interferon regulatory factor 3 (IRF3), a regulator of type I interferons (IFN-Is), including interferon alpha (IFNα) and beta (IFNβ), further increasing the mitophagy activity and suppressing the IFN-I-mediated metabolic inflammation pathway. Meanwhile, ATF5 is translationally expressed by GCN2-dependent eIF2α phosphorylation and regulates metabolism, cell growth with ATF4 by conjointly activating fibroblast growth factor 21 (FGF21), and a pressure sensor in the liver that regulates metabolism via binding to its promoter amino acid response elements site during periods of nutrient restriction [50]. All indicate that ATF5 participates in the restoration of hepatic metabolic homeostasis in liver cells.
2.3. Reverse ATF5 Regulation
In addition to factors that enhance the transcriptional activities of ATF5, several known repressors of ATF5 exist (Figure 3). The ATF5–nucleophosmin 1 (NPM1) protein interaction promotes ATF5 degradation through proteasome- and caspase-dependent pathways in HCC cells. This replaces the interaction of the ATF5 protein with heat-shock protein 70 (HSP70), which maintains high levels of ATF5 expression by preventing its degradation in glioma cells [51][52].
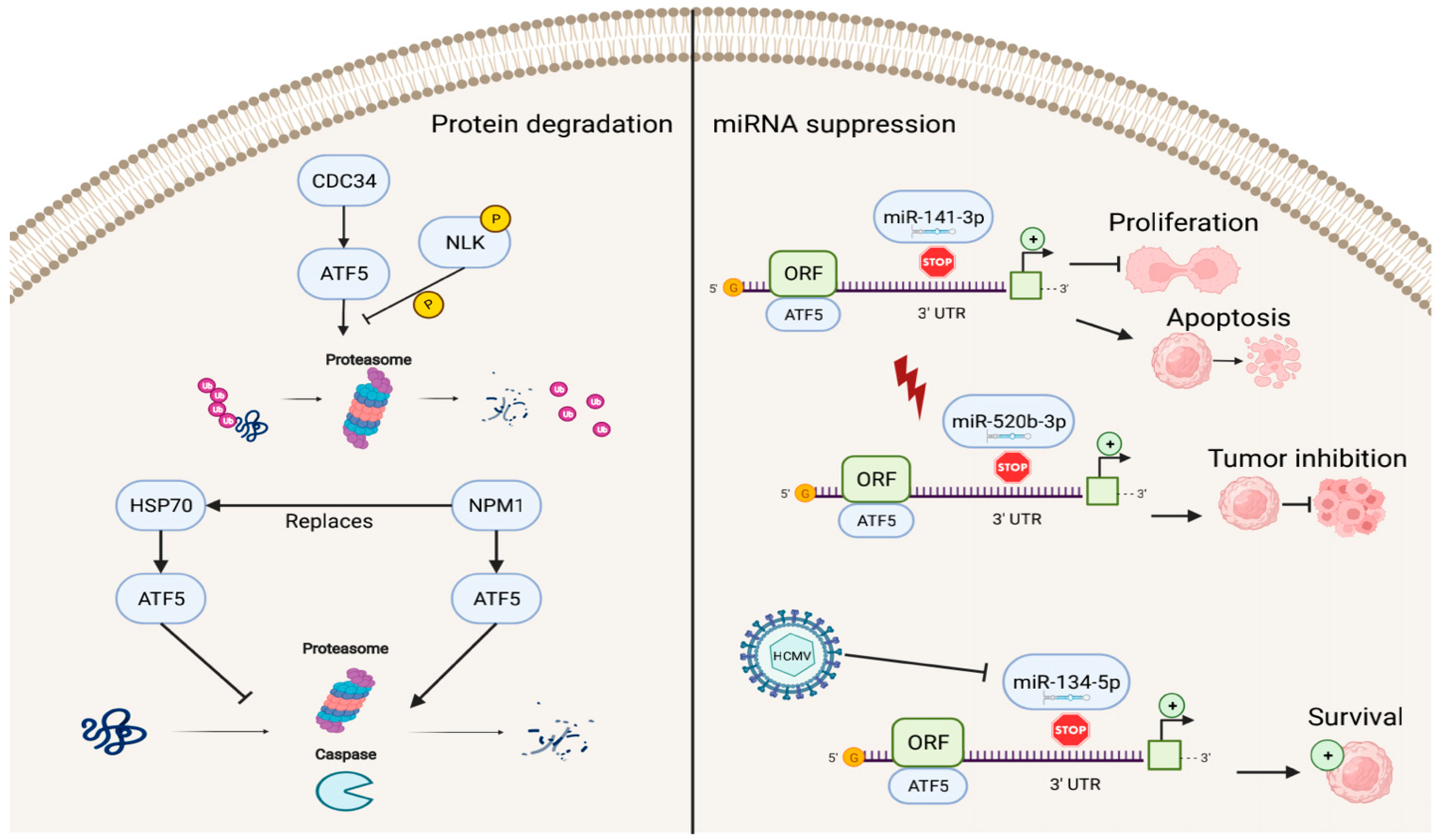
Figure 3. Reverse regulation of ATF5 expression. ATF5 can be degraded by cell division cycle 34 (CDC34), nucleophosmin 1 (NPM1), and miRNAs (miR-141-3p, miR-520b-3p, and miR-134-5p), resulting in inhibition of cell proliferation, induction of apoptosis, and decreased survival of cancer cells.
ATF5 post-transcriptional expression can also be suppressed by miRNAs, including miR-141-3p, miR-520b-3p, and miR-134-5p, which bind to the 3′ UTR site of ATF5 mRNA and regulate ATF5 expression. In this process, the down-regulation of ATF5 expression by miR-141-3p inhibits cell proliferation and promotes cell apoptosis in gliomas [53]. miR-520b-3p can suppress ATF5 expression levels and play a positive role in tumor inhibition during diverse stress conditions in ATF5-oncogenic cancer cells; however, the specific mechanisms of miR-520b-3p-induced ATF5 inhibition are not fully understood and require further investigation [54]. Furthermore, HCMV infection can trigger miR-134-5p down-regulation, which is the negative regulator of ATF5 expression, thus, resulting in high levels of ATF5 and increased glioma cell survival [55].
References
- Mason, J.L.; Angelastro, J.M.; Ignatova, T.N.; Kukekov, V.G.; Lin, G.; Greene, L.A.; Goldman, J.E. ATF5 regulates the proliferation and differentiation of oligodendrocytes. Mol. Cell. Neurosci. 2005, 29, 372–380.
- Angelastro, J.M.; Ignatova, T.N.; Kukekov, V.G.; Steindler, D.A.; Stengren, G.B.; Mendelsohn, C.; Greene, L.A. Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J. Neurosci. 2003, 23, 4590–4600.
- Angelastro, J.M.; Mason, J.L.; Ignatova, T.N.; Kukekov, V.G.; Stengren, G.B.; Goldman, J.E.; Greene, L.A. Downregulation of activating transcription factor 5 is required for differentiation of neural progenitor cells into astrocytes. J. Neurosci. 2005, 25, 3889–3899.
- Lee, H.Y.; Angelastro, J.M.; Kenney, A.M.; Mason, C.A.; Greene, L.A. Reciprocal actions of ATF5 and Shh in proliferation of cerebellar granule neuron progenitor cells. Dev. Neurobiol. 2012, 72, 789–804.
- Umemura, M.; Kaneko, Y.; Tanabe, R.; Takahashi, Y. ATF5 deficiency causes abnormal cortical development. Sci. Rep. 2021, 11, 7295.
- Umemura, M.; Ogura, T.; Matsuzaki, A.; Nakano, H.; Takao, K.; Miyakawa, T.; Takahashi, Y. Comprehensive Behavioral Analysis of Activating Transcription Factor 5-Deficient Mice. Front. Behav. Neurosci. 2017, 11, 125.
- Umemura, M.; Tsunematsu, K.; Shimizu, Y.I.; Nakano, H.; Takahashi, S.; Higashiura, Y.; Okabe, M.; Takahashi, Y. Activating transcription factor 5 is required for mouse olfactory bulb development via interneuron. Biosci. Biotechnol. Biochem. 2015, 79, 1082–1089.
- Wang, S.-Z.; Ou, J.; Zhu, L.J.; Green, M.R. Transcription factor ATF5 is required for terminal differentiation and survival of olfactory sensory neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 18589–18594.
- Zhao, Y.; Zhang, Y.D.; Zhang, Y.Y.; Qian, S.W.; Zhang, Z.C.; Li, S.F.; Guo, L.; Liu, Y.; Wen, B.; Lei, Q.Y.; et al. p300-dependent acetylation of activating transcription factor 5 enhances C/EBPbeta transactivation of C/EBPalpha during 3T3-L1 differentiation. Mol. Cell. Biol. 2014, 34, 315–324.
- Leong, D.T.; Abraham, M.C.; Gupta, A.; Lim, T.C.; Chew, F.T.; Hutmacher, D.W. ATF5, a possible regulator of osteogenic differentiation in human adipose-derived stem cells. J. Cell. Biochem. 2012, 113, 2744–2753.
- Du, Y.; Wang, J.; Jia, J.; Song, N.; Xiang, C.; Xu, J.; Hou, Z.; Su, X.; Liu, B.; Jiang, T.; et al. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell 2014, 14, 394–403.
- Zhang, Q.; Huang, Z.; Zuo, H.; Lin, Y.; Xiao, Y.; Yan, Y.; Cui, Y.; Lin, C.; Pei, F.; Chen, Z.; et al. Chromatin Accessibility Predetermines Odontoblast Terminal Differentiation. Front. Cell Dev. Biol. 2021, 9, 769193.
- Zhang, S.; Chen, J.J. Requirement of activating transcription factor 5 for murine fetal liver erythropoiesis. Br. J. Haematol. 2020, 188, 582–585.
- Madarampalli, B.; Yuan, Y.; Liu, D.; Lengel, K.; Xu, Y.; Li, G.; Yang, J.; Liu, X.; Lu, Z.; Liu, D.X. ATF5 Connects the Pericentriolar Materials to the Proximal End of the Mother Centriole. Cell 2015, 162, 580–592.
- Yuan, Y.; Gaither, K.; Kim, E.; Liu, E.; Hu, M.; Lengel, K.; Qian, D.; Xu, Y.; Wang, B.; Knipprath, H.; et al. SUMO2/3 modification of activating transcription factor 5 (ATF5) controls its dynamic translocation at the centrosome. J. Biol. Chem. 2018, 293, 2939–2948.
- Zhou, D.; Palam, L.R.; Jiang, L.; Narasimhan, J.; Staschke, K.A.; Wek, R.C. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 2008, 283, 7064–7073.
- Pei, Z.; Zhang, X.; Ji, C.; Liu, S.M.; Wang, J. Transcriptomic and functional pathways analysis of ascorbate-induced cytotoxicity and resistance of Burkitt lymphoma. Oncotarget 2016, 7, 63950–63959.
- Torres-Peraza, J.F.; Engel, T.; Martin-Ibanez, R.; Sanz-Rodriguez, A.; Fernandez-Fernandez, M.R.; Esgleas, M.; Canals, J.M.; Henshall, D.C.; Lucas, J.J. Protective neuronal induction of ATF5 in endoplasmic reticulum stress induced by status epilepticus. Brain 2013, 136, 1161–1176.
- Yasuda, M.; Tanaka, Y.; Ryu, M.; Tsuda, S.; Nakazawa, T. RNA sequence reveals mouse retinal transcriptome changes early after axonal injury. PLoS ONE 2014, 9, e93258.
- Juliana, C.A.; Yang, J.; Rozo, A.V.; Good, A.; Groff, D.N.; Wang, S.Z.; Green, M.R.; Stoffers, D.A. ATF5 regulates beta-cell survival during stress. Proc. Natl. Acad. Sci. USA 2017, 114, 1341–1346.
- Izumi, S.; Saito, A.; Kanemoto, S.; Kawasaki, N.; Asada, R.; Iwamoto, H.; Oki, M.; Miyagi, H.; Ochi, M.; Imaizumi, K. The endoplasmic reticulum stress transducer BBF2H7 suppresses apoptosis by activating the ATF5-MCL1 pathway in growth plate cartilage. J. Biol. Chem. 2012, 287, 36190–36200.
- Wang, H.; Lin, G.; Zhang, Z. ATF5 promotes cell survival through transcriptional activation of Hsp27 in H9c2 cells. Cell Biol. Int. 2007, 31, 1309–1315.
- Deng, P.; Haynes, C.M. Mitochondrial dysfunction in cancer: Potential roles of ATF5 and the mitochondrial UPR. Semin. Cancer Biol. 2017, 47, 43–49.
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043.
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855.
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366.
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701.
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478.
- Zhang, B.; Tan, Y.; Zhang, Z.; Feng, P.; Ding, W.; Wang, Q.; Liang, H.; Duan, W.; Wang, X.; Yu, S.; et al. Novel PGC-1alpha/ATF5 Axis Partly Activates UPR(mt) and Mediates Cardioprotective Role of Tetrahydrocurcumin in Pathological Cardiac Hypertrophy. Oxid. Med. Cell. Longev. 2020, 2020, 9187065.
- Wen, S.; Unuma, K.; Funakoshi, T.; Aki, T.; Uemura, K. Altered cardiac mitochondrial dynamics and biogenesis in rat after short-term cocaine administration. Sci. Rep. 2021, 11, 24129.
- Nandi, S.S.; Maresh, G.M.; Patel, K.P. Decreased Mitochondrial Unfolded Protein Response (UPRmt) in HFpEF. FASEB J. 2022, 36.
- Sheng, Z.; Li, L.; Zhu, L.J.; Smith, T.W.; Demers, A.; Ross, A.H.; Moser, R.P.; Green, M.R. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat. Med. 2010, 16, 671–677.
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848.
- Hua, Z.Y.; Hansen, J.N.; He, M.; Dai, S.K.; Choi, Y.; Fulton, M.D.; Lloyd, S.M.; Szemes, M.; Sen, J.; Ding, H.F.; et al. PRMT1 promotes neuroblastoma cell survival through ATF5. Oncogenesis 2020, 9, 50.
- Hu, M.; Li, H.; Xie, H.; Fan, M.; Wang, J.; Zhang, N.; Ma, J.; Che, S. ELF1 Transcription Factor Enhances the Progression of Glioma via ATF5 promoter. ACS Chem. Neurosci. 2021, 12, 1252–1261.
- Zhou, J.; Tian, H.; Zhi, X.; Xiao, Z.; Chen, T.; Yuan, H.; Chen, Q.; Chen, M.; Yang, J.; Zhou, Q.; et al. Activating transcription factor 5 (ATF5) promotes tumorigenic capability and activates the Wnt/b-catenin pathway in bladder cancer. Cancer Cell Int. 2021, 21, 660.
- He, F.; Xiao, H.; Cai, Y.; Zhang, N. ATF5 and HIF1alpha cooperatively activate HIF1 signaling pathway in esophageal cancer. Cell Commun. Signal. 2021, 19, 53.
- Nukuda, A.; Endoh, H.; Yasuda, M.; Mizutani, T.; Kawabata, K.; Haga, H. Role of ATF5 in the invasive potential of diverse human cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 474, 509–514.
- Heino, J. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol. 2000, 19, 319–323.
- Gho, J.W.; Ip, W.K.; Chan, K.Y.; Law, P.T.; Lai, P.B.; Wong, N. Re-expression of transcription factor ATF5 in hepatocellular carcinoma induces G2-M arrest. Cancer Res. 2008, 68, 6743–6751.
- Wei, Y.; Jiang, J.; Sun, M.; Chen, X.; Wang, H.; Gu, J. ATF5 increases cisplatin-induced apoptosis through up-regulation of cyclin D3 transcription in HeLa cells. Biochem. Biophys. Res. Commun. 2006, 339, 591–596.
- Liu, W.; Sun, M.; Jiang, J.; Shen, X.; Sun, Q.; Liu, W.; Shen, H.; Gu, J. Cyclin D3 interacts with human activating transcription factor 5 and potentiates its transcription activity. Biochem. Biophys. Res. Commun. 2004, 321, 954–960.
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490.
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065.
- Juliana, C.A.; Yang, J.; Cannon, C.E.; Good, A.L.; Haemmerle, M.W.; Stoffers, D.A. A PDX1-ATF transcriptional complex governs beta cell survival during stress. Mol. Metab. 2018, 17, 39–48.
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273.
- Abe, T.; Kojima, M.; Akanuma, S.; Iwashita, H.; Yamazaki, T.; Okuyama, R.; Ichikawa, K.; Umemura, M.; Nakano, H.; Takahashi, S.; et al. N-terminal hydrophobic amino acids of activating transcription factor 5 (ATF5) protein confer interleukin 1beta (IL-1beta)-induced stabilization. J. Biol. Chem. 2014, 289, 3888–3900.
- Lei, Z.; Yang, L.; Yang, Y.; Yang, J.; Niu, Z.; Zhang, X.; Song, Q.; Lei, Y.; Wu, H.; Guo, J. Activation of Wnt/beta-catenin pathway causes insulin resistance and increases lipogenesis in HepG2 cells via regulation of endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2020, 526, 764–771.
- Liou, Y.H.; Personnaz, J.; Jacobi, D.; Knudsen, N.H.; Chalom, M.M.; Starost, K.A.; Nnah, I.C.; Lee, C.H. Hepatic Fis1 regulates mitochondrial integrated stress response and improves metabolic homeostasis. JCI Insight 2022, 7, e150041.
- Laeger, T.; Henagan, T.M.; Albarado, D.C.; Redman, L.M.; Bray, G.A.; Noland, R.C.; Münzberg, H.; Hutson, S.M.; Gettys, T.W.; Schwartz, M.W.; et al. FGF21 is an endocrine signal of protein restriction. J. Clin. Investig. 2014, 124, 3913–3922.
- Liu, X.; Liu, D.; Qian, D.; Dai, J.; An, Y.; Jiang, S.; Stanley, B.; Yang, J.; Wang, B.; Liu, X.; et al. Nucleophosmin (NPM1/B23) interacts with activating transcription factor 5 (ATF5) protein and promotes proteasome- and caspase-dependent ATF5 degradation in hepatocellular carcinoma cells. J. Biol. Chem. 2012, 287, 19599–19609.
- Li, G.; Xu, Y.; Guan, D.; Liu, Z.; Liu, D.X. HSP70 protein promotes survival of C6 and U87 glioma cells by inhibition of ATF5 degradation. J. Biol. Chem. 2011, 286, 20251–20259.
- Wang, M.; Hu, M.; Li, Z.; Qian, D.; Wang, B.; Liu, D.X. miR-141-3p functions as a tumor suppressor modulating activating transcription factor 5 in glioma. Biochem. Biophys. Res. Commun. 2017, 490, 1260–1267.
- Gaither, K.A.; Watson, C.J.W.; Madarampalli, B.; Lazarus, P. Expression of activating transcription factor 5 (ATF5) is mediated by microRNA-520b-3p under diverse cellular stress in cancer cells. PLoS ONE 2020, 15, e0225044.
- Hu, M.; Yu, B.; Zhang, B.; Wang, B.; Qian, D.; Li, H.; Ma, J.; Liu, D.X. Human Cytomegalovirus Infection Activates Glioma Activating Transcription Factor 5 via microRNA in a Stress-Induced Manner. ACS Chem. Neurosci. 2021, 12, 3947–3956.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
2 times
(View History)
Update Date:
27 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No