Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Romelia Pop | -- | 4894 | 2022-07-26 18:04:45 | | | |
| 2 | Rita Xu | Meta information modification | 4894 | 2022-07-27 03:21:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pop, R.; Tăbăran, A.; Ungur, A.P.; Negoescu, A.; Cătoi, C. Helicobacter Pylori-Induced Gastric Infections. Encyclopedia. Available online: https://encyclopedia.pub/entry/25534 (accessed on 08 August 2026).
Pop R, Tăbăran A, Ungur AP, Negoescu A, Cătoi C. Helicobacter Pylori-Induced Gastric Infections. Encyclopedia. Available at: https://encyclopedia.pub/entry/25534. Accessed August 08, 2026.
Pop, Romelia, Alexandru-Flaviu Tăbăran, Andrei Paul Ungur, Andrada Negoescu, Cornel Cătoi. "Helicobacter Pylori-Induced Gastric Infections" Encyclopedia, https://encyclopedia.pub/entry/25534 (accessed August 08, 2026).
Pop, R., Tăbăran, A., Ungur, A.P., Negoescu, A., & Cătoi, C. (2022, July 26). Helicobacter Pylori-Induced Gastric Infections. In Encyclopedia. https://encyclopedia.pub/entry/25534
Pop, Romelia, et al. "Helicobacter Pylori-Induced Gastric Infections." Encyclopedia. Web. 26 July, 2022.
Copy Citation
Helicobacter pylori is the first formally recognized bacterial carcinogen and the most important single digestive pathogen responsible for the induction of gastroduodenal diseases such as gastritis, peptic ulcer, and, finally, gastric neoplasia. The recently reported high rates of antimicrobial drug resistance hamper the current therapies of H. pylori, with therapeutic failure reaching up to 40% of patients.
silver nanoparticles
Helicobacter pylori
1. Introduction
Helicobacter pylori, a spiral-shaped or coccoid [1] Gram-negative bacterium is the most common infectious agent of gastric diseases worldwide. H. pylori infect 30–70% of the world’s population, being one of the leading causes of gastrointestinal diseases, producing functional dyspepsia, acute and chronic gastritis, and peptic ulcer in up to 20% of infected cases. Importantly, following H. pylori infection, there is a 3–6 times higher risk of developing aggressive gastric cancers, including gastric adenocarcinoma and mucosa-associated lymphoid lymphoma (MALT lymphoma) [2]. In this context, H. pylori is the single bacterium classified as a group I carcinogen by the World Health Organization (WHO) [3]. In this context, the eradication of H. pylori infection is indicated as the first choice of treatment for several of these gastric malignancies, including MALT lymphoma [4]. In addition to the well-established gastric pathologies, recently, a considerable amount of evidence has linked H. pylori infection with several extragastric diseases within the nervous, cardiovascular, and immune systems [5], such as dementia, Alzheimer’s disease [6], Parkinson’s disease [7], Guillain–Barré syndrome [8], iron deficiency anemia/aregenerative anemia [9], coronary atherosclerosis [10], and nonalcoholic steatosis [11]. The H. pylori infection is typically acquired in early childhood, either by fecal–oral or oral–oral route, and in the absence of effective treatment, it lasts a lifetime [12][13]. This indicates a strong adaptation to the biological niche represented by the mucous layer covering the gastric epithelial cells. The resistance mechanism of H. pylori within the low-pH environment of the stomach is particularly complex, involves the expression of several genes considered key pathogenic factors (as CagA, VacA, BabA, urease, etc.), and determines several particularities in the currently employed therapies, such as simultaneous administration of several active molecules (typically at least two antibiotics and a proton pump inhibitor) and long-term treatments (lasting at least 2 weeks) [14].
1.1. Current Treatment Protocols for H. pylori Infection and Emerging Antibiotic-Resistant Strains
The first-line option for H. pylori infection is represented by a standard triple therapy (STT) consisting of two antibiotics, clarithromycin and amoxicillin or metronidazole in combination with a proton pump inhibitor (PPI). The second-line option for H. pylori treatment is represented by bismuth-based quadruple therapy. If the first- and second-line therapy fails, triple therapies (based on levofloxacin and/or rifabutin in combination with amoxicillin) can be an option. Furthermore, sequential therapy including initially dual administration of a proton pump inhibitor (PPI) plus amoxicillin followed by a triple therapy including a PPI, clarithromycin, and tinidazole was developed as a therapeutic alternative [15].
Successful treatment of H. pylori-induced gastritis has become a challenge in recent years [16] due to several factors, including the emergence of new antibiotic-resistant strains (e.g., up to 72.5%-resistant for amoxicillin, 58.8% for clarithromycin, and 34.9% for metronidazole) [17][18] and poor patient compliance. In the “antibiotic resistance era”, the low eradication rate and suboptimal results of the classical therapy are reported in both children and adults, with some studies showing therapeutic failure in up to 40% of the children and 38% of the adults subjected to the standard triple therapy for H. pylori infections in China [19][20]. Bismuth-based quadruple therapy has been proven to have better results regarding the treatment of H. pylori. Luminal bismuth is responsible for the action of bismuth salts within the upper gastrointestinal system. Inhibiting a variety of enzymes, ATP generation, and the bacteria’s adhesion to the stomach mucosa are only a few of the ways that bismuth has a direct bactericidal effect on H. pylori. Additionally, bismuth promotes the release of prostaglandin, epidermal growth factor, and bicarbonate, which are mucosa-protective substances that aid in the healing of ulcers. No reports of bismuth resistance exist as of yet. The suboptimal results in the classical therapies merged with the need for the development of new treatment agents for H. pylori-induced gastritis [19]. Based on the high rates of antibiotic resistance and recent epidemiological evidence, in 2017, the WHO listed H. pylori among the sixteen antibiotic-resistant bacteria ranked as high-priority bacteria that pose the greatest threat to human health worldwide [21].
1.2. Potential Use of AgNPs as a Complementary or Alternative Therapy for H. pylori in the Context of Antibiotic Resistance
Pathogens that are resistant to older antimicrobials and compounds that are regarded last-resort treatments have continued to emerge as a result of antibiotic use. Therapy effectiveness is significantly harmed by antimicrobial resistance, and the risk of cross-infection in hospitals has been on the rise. Resistance causes ineffective empirical therapy, a delay in beginning treatment, and the use of less effective, more harmful, and more expensive medications [22][23][24].
Growing rates of antibiotic resistance of H. pylori are reported worldwide. This is highlighted by the overall low resistance rates reported in the first decade of 2000 [25]. In response to this high rate of resistance to antibiotics used in the classical therapy, recently, the World Health Organization (WHO) designated the clarithromycin-resistant strains of H. pylori as a high priority for antibiotic research and the development of alternative solutions. A study conducted from April 2008 to June 2009 in 18 European countries showed that rates of resistance for adults were 34.9% for metronidazole, 14.1% for levofloxacin, and 17.5% for clarithromycin [18]. In the period from 2008 to 2017, there was another study conducted by Megruad et al. [26] in the same 18 European states that showed the resistance rates were 38.9% for metronidazole, 15.8% for levofloxacin, and 21.4% for clarithromycin.
Antibiotic resistance has increased worldwide, which seriously hampers the eradication rate of the frequent chronic infection. Areas covered: H. pylori MDR rates are discussed, mostly in the recent articles published since 2015. Present approaches and future directions to counteract MDR are outlined. Some studies have shown that probiotics have an inhibitory effect on H. pylori but other studies have proven that their effect is limited [27][28]. Approximately 20% of patients do not respond positively to the recommended treatment regimen. Inadequate patient compliance, obesity, smoking, reinfection, genetic polymorphisms in CYP2C19, poor patient maintenance, improper alimentary regimen, antibiotic resistance, disease entities associated with H. pylori infection, antibiotic degradation by the acidic stomach environment, and pharmacological activity of the prescribed drugs are the major causes of treatment failure [3][29][30]. The main cause of eradication failure is represented by the resistance to antibiotics [31].
Because of the major need to avoid antibiotic resistance and the high infection rate of H. pylori, it is necessary to develop an effective alternative to the typical antibiotic therapy. The recent studies have shown an efficient antimicrobial effect of metal nanoparticles using their potential in the medical field [32][33][34]. It is known that metallic nanoparticles were synthesized centuries ago because of their intrinsic physical and chemical characteristics, especially their optoelectronic properties and surface plasma resonance [35]. Silver-based nanoparticles have been proven to be effective against multiple microorganisms, such as bacteria, viruses, and bacterial biofilm. Due to the attachment on the cell surface, silver nanoparticles interact with bacteria, changing the wall structure and causing damage to cell functions, increasing their permeability by pore formation and affecting the respiratory enzymes. These actions eventually lead to cell death [36]. One of the major public health problems caused by the infection with multidrug-resistant bacteria is morbidity and mortality which cause the need to implement measures to control the infection.
The development of new antibacterial drugs against H. pylori and identification of new drug targets for the diseases caused by persistent H. pylori infections must first take into account the complex diseases with complicated pathogenesis and the high adaptability of H. pylori to the gastric environment. Next, researchers present briefly the main pathologies induced by persistent H. pylori gastric infections, highlighting the potential usage of AgNPs in the therapy and prevention of these infections in the context of pathogenicity.
In addition to the usage of nanoparticles as direct antibacterial agents, currently, the nanotechnology of advanced encapsulation strategies enables novel material–cell interactions, such as reversible encapsulation of living-single cells [37], offering fascinating biomedical opportunities in drug delivery and transplantation [38] Drug delivery systems are a relatively young but quickly developing technology. In these systems, therapeutically effective drugs or imaging molecules are delivered to the targets using nanoscale materials. These carrier systems, which improve medication formulation, targeting, and controlled release, are fundamental to personalized medicine and depend heavily on nanoparticles and nanostructured materials. Such systems can deliver medicines at a predetermined pace and in a preplanned manner to a specific region; as a result, the drug’s bioavailability is increased and its negative effects are decreased. Drugs may be placed onto nanoparticles during manufacturing or may be physically or chemically adsorbed into the surface of the particles using various adsorption techniques [39][40][41].
2. The Pathology of Helicobacter pylori Infection: From Inflammation to Gastric Neoplasia
The pathogenic process of H. pylori is particularly complex and consists of three important steps: (1) colonization, (2) immune escape, and (3) disease induction (Figure 1). The bacterium colonizes the stomach, settling in the deep portions of the gelatinous mucous layer lining the gastric mucosa and between the mucous layer and the apical surface of the epithelial cells of the gastric mucosa. The gastric mucosa is well-protected against bacterial infection. H. pylori is well-adapted to this ecological niche, having unique characteristics that allow it to enter mucus, attach to epithelial cells, evoke the immune response and, consequently, persistent colonization and transmission. The colonization of the mucosa is due to a set of aggressive enzymes (urease, mucinase, peptidases, etc.) that render it pathogenic, the bacterium being located under the mucous layer, between the epithelial cells, and around the gastric crypts. Bacterial phospholipases destroy the bilipid layer of the gastroduodenal epithelial cell membrane. Being a bacterium with very high mobility, it has flagella that allow it to easily cross the viscous layer of the mucosa to reach the levels of gastric mucosa cells where pH is almost neutral. Colonization of the mucosa would not be possible if the microbe did not protect itself from the acidic environment of the stomach. The next aggression factor is urease, a surface enzyme in the bacterial body which hydrolyzes urea, causing an increased amount of ammonia, which surrounds the bacterial body like a cloud. The ammonia formed is associated with hydrochloric acid forming extremely toxic products: hydroxylamine and monochloramine [42]. The following factors participate in the induction of gastroduodenal diseases: adaptation factors, bacterial survival in the host environment, and virulence factors (Table 1). Pathogenic factors can be exemplified by the protein products of CagA, VacA, IceA, and BabA genes [43].
Urease produced by H. pylori catalyzes the hydrolysis of urea into ammonia and carbon dioxide, which are necessary for gastric colonization and the protection of the bacillus from the effects of gastric acid. Hydroxide ions generated by the reaction of water with ammonia can contribute to damage to the gastric epithelial mucosa, and surface proteins are chemotactic for polymorphonuclear cells and monocytes. It also secretes platelet-activating factors, activates monocytes, produces superoxide, interleukin 1, tumor necrosis factor, protease, and phospholipases, which degrade the glycoprotein–lipid complex of the mucous layer. The first pathogenicity factor described was the CagA gene (cytotoxin-associated gene), which is part of the pathogenicity island (PAI) [44]. The protein secreted by this gene hitting the epithelial cells of the gastric mucosa induces their modification, damaging their cytoskeleton, and induces the synthesis of protein kinases, nuclear factor NF-kB, proinflammatory interleukins 1, 6, 8, chemotactic monocyte protein, tumor necrosis factor (TNF) [45]. Interleukin 8 is a powerful factor that accelerates the movement of neutrophils, and the chemotactic monocyte protein accelerates the movement of monocytes to the source, with the infiltration of the gastroduodenal mucosa with inflammatory cells: neutrophils and monocytes. This explains the predominance of intensely inflammatory reactions in people with pathogenic CagA+ strains compared to CagA strains. Some authors believe that CagA+ H. pylori strains inhibit phagocytosis, compromising the host’s immune response [46].

Figure 1. H. pylori infection pathogenicity and inflammatory response (PLA—phospholipases; Dupg—duodenal ulcer-promoting gene A; CagA—cytotoxin-associated gene A; BabA—blood group antigen-binding adhesin; VacA—vacuolating cytotoxin A; CXCL—chemokine ligand; VCAM—vascular cell adhesion molecules; ICAM—intercellular cell adhesion molecules). Created using BioRender (adapted from [47][48]).
Patients infected with CagA+ H. pylori have a 12 times higher risk of developing intestinal metaplasia of the gastroduodenal mucosa, which explains the possibility of further development of gastric cancer [49]. Another pathogenicity factor is VacA (vacuolating cytotoxin gene) which secretes a cytotoxic protein that damages epithelial cells, forming pores in the cytoplasmic membrane, increasing their permeability to the formation of vacuoles inside the cell, so it is a gene encoding the cytotoxin inducing vacuolization [50]. The regeneration of the ulcer defect depends on the balance between the processes of proliferation and apoptosis. The chronic ulcerative defect is caused by numerous factors, including the high apoptotic activity of epithelial cells. The predominance of apoptosis over-proliferation can be the cause of gastroduodenal ulcers with persistent evolution and prolonged scarring. Conversely, the predominance of proliferative processes over apoptosis increases the number of epithelial cells whose nucleus contains damaged, immature DNA, and thus the accumulation of mutations, which explains the carcinogenic mechanism of H. pylori. In response to long-term H. pylori infection and permanent damage to the gastroduodenal mucosa, cell proliferation processes intensify [51]. Cytotoxic strains CagA and VacA are characterized by greater stimulation of the proliferation processes. It was found that after the eradication of H. pylori in the antral region, the proliferation processes are reduced considerably and early, while in the gastric body, the normalization of the imbalance of the proliferation processes takes place much later. Some authors have shown in vivo that CagA-positive H. pylori strains (CagA+ H. pylori) stimulate proliferation but, unlike H. pylori-negative strains (CagA– H. pylori), inhibit apoptosis, having a lower apoptotic index [52]. In 1998, R. Peek et al. [50] identified the IceA gene (induced by contact with the epithelium). This gene exists in two alleles, IceA1 and IceA2; the proteins secreted by this gene ensure adhesion to epithelial cells of the gastroduodenal mucosa. According to some authors, the presence of the IceA1 gene determines the development of gastric ulcers and the presence of neutrophil infiltration of the gastroduodenal mucosa [53]. Another factor in the adhesion of H. pylori to the gastroduodenal epithelium is the product of the BabA gene (blood group antigen-binding adhesin) which is a mediator of H. pylori adhesion to the Lewis antigen system, which ensures the persistence of the bacterium in the gastroduodenal area. The presence of this gene is considered a marker of the development of complications [52]. H. pylori cause a continuous inflammation of the gastric mucosa in all infected people. This inflammatory response initially consists of the recruitment of neutrophils, followed by T and B lymphocytes, plasma cells, and macrophages, with alteration of the epithelial layer (Figure 1). Differences in the immune response reflect the diversity of clinical manifestations of H. pylori infection [54]. H. pylori can live for years and sustain an intense inflammatory process because of their toxins such as vacuolating cytotoxin encoded by the VacA gene and immunogenic protein CagA encoded by the CagA gene (cytotoxin-associated gene, found only in the strains that produce an aggressive form of gastroduodenal diseases). The last one is located in the H. pylori pathogenicity island (PAI) [55][56][57]. To survive in a low-pH environment, H. pylori produce urease (the main virulence factor used for diagnosis), an enzyme that helps in the colonization of gastric mucosa. Urease has a major role in the neutralization of the acid fluid by catalyzing the degradation of urea to carbon dioxide and ammonia and raising the pH of the environment. It was showed that for the production of urease, H. pylori uses at least seven genes: ureEFGH encodes the accessor proteins that are in control of integrating nickel in the active center of the enzyme while the ureAB gene encodes two genes of the enzyme and ureI encodes the proteins that are in charge of urea transport into the target cell. All strains of H. pylori contain two phospholipases (A and C). The role of these two enzymes is to destroy the mucous layer [56].
Table 1. Synopsis of the main H. pylori virulence factors and their pathogenic mechanism [48][55][58][59].
| Bacterial Element | Virulence Factors | Mechanism of Bacterial Virulence |
|---|---|---|
| Flagella | Flagellum | Bacterial motility |
| Leukocyte chemotaxis | ||
| Biofilm formation | ||
| Inflammation and immune response | ||
| Adhesins | Blood group antigen-binding adhesin (BabA) | Adherence to gastric epithelial cells |
| Toxins delivery | ||
| Increasing inflammatory responses | ||
| Sialic acid-binding adhesin (SabA) | Neutrophil activation | |
| Colonization | ||
| Oxidative stress | ||
| Adherence-associated lipoproteins A and B | Adherence to gastric epithelial cells | |
| Colonization | ||
| Biofilm formation | ||
| Release of proinflammatory factors | ||
| LacdiNAc-specific adhesin (LabA) | Adherence to gastric epithelial cells | |
| Enzymes | Urease | Protection from acidity |
| Colonization | ||
| Bacterial nutrition | ||
| Control of the host immune response | ||
| Platelet activation | ||
| Angiogenesis | ||
| Catalase | DNA damage and mutagenesis | |
| Induction of inflammation | ||
| Survival of phagocytosis | ||
| Superoxidase dismutase (SOD) | Gastric colonization | |
| Protection from reactive oxygen species | ||
| Inhibition of the synthesis of cytokines | ||
| Activation of macrophages | ||
| Arginase | Apoptosis | |
| Protection from acidity | ||
| Dysregulation of the immune response by inhibition of T and B cells production | ||
| Macrophage apoptosis | ||
| Phospholipases (PLAs) | Degradation of lipids | |
| Lysis of the mucous layer | ||
| Chronic inflammation induction | ||
| Gastric colonization | ||
| Cholesteryl ꭤ-glucosyltransferase | Protection from immune responses and phagocytosis | |
| Secretion of proinflammatory factors | ||
| Antibiotic resistance | ||
| Bacterial growth | ||
| ɤ-glutamyl-transpeptidase (GGT) | Stimulation of the release of SOR | |
| Apoptosis and necrosis | ||
| Induction of the release of proinflammatory factors | ||
| DNA damage | ||
| Decrease in cell viability | ||
| High-temperature-requirement serine protease A (HTRA) | Damage of the gastric epithelium | |
| Bacterial mobility | ||
| Proteins | Cytotoxin-associated gene A (CagA) | Induction of the inflammatory response |
| Bacterial motility | ||
| Activation of fibroblasts | ||
| Oncogenesis (by dysregulation of the RUNX3, ASPP2, CDX1, and AFADIN genes) | ||
| Decrease in microRNA-134, PDCD4, GSK-3 | ||
| Tumor progression by induction of cancer stem cell-like characteristics | ||
| Vacuolating cytotoxin A (VacA) | Induction of autophagy and formation of autophagosomes | |
| Induction of cellular apoptosis and necrosis | ||
| Dysregulation of the immune response by inhibition of T and B cells production | ||
| Outer inflammatory protein A (OipA) | Induction of apoptosis | |
| Induction of the release of proinflammatory factors | ||
| CagA delivery Stimulation of the microRNA-30b level |
||
| Outer membrane protein Q (HopQ) | Bacterial adherence to gastric epithelial cells | |
| Protection from gastric acidity | ||
| Induction of the release of proinflammatory factors | ||
| Inhibition of the immune response | ||
| Outer membrane protein Z | Adherence to gastric epithelial cells | |
| Increase in gastric secretion | ||
| Neutrophil-activating protein (NAP) | Stimulation of neutrophils adhesion to gastric epithelial cells | |
| Heat shock proteins (Hsps) | Maintenance of intact functional and structural characteristics of cellular proteins | |
| Chronic inflammation and angiogenesis | ||
| Adherence to gastric epithelial cells | ||
| Induction of apoptosis autophagy | ||
| Urease activation | ||
| Gastric tumor cells migrations | ||
| Toxins | Lipopolysaccharide (LPS) | Stimulation of the inflammatory response by neutrophil activation |
| Induction of bacterial protection | ||
| Impairment of the gastric mucosa mucus production | ||
| Others | Lewis antigens | Protection from host defense |
| Bacterial protection | ||
| Adhesive proprieties | ||
| Duodenal ulcer promoting gene A (DupA) | Increase in the inflammatory response | |
| Induction of apoptosis (intrinsic pathway) | ||
| Bacterial resistance to the acidic microenvironment |
There are several gastric and extragastric pathologic effects of the chronic H. pylori infection. To hypostatize which point of H. pylori infection would benefit from AgNP therapy, in the next section, the main gastric lesions (including inflammatory and neoplastic changes) and extragastric pathologies are detailed.
2.1. Gastric Inflammation Induced by H. pylori Infection
All H. pylori infections start with acute gastritis which can persist ofr over 15 days after colonization. In a few cases healing moves into the chronic form. Tissue changes are characterized by degradation of the surface epithelium, the presence of numerous neutrophils located inside the epithelium, in the lamina propria, or aggregated in glandular lumens (cryptic abscesses). Acute gastritis induced by H. pylori is associated with nodular gastritis, follicular gastritis (Figure 2), hemorrhagic gastritis, lymphocytic gastritis, granulomatous gastritis, and hypertrophic gastritis. These lesions can be reversible [60][61].
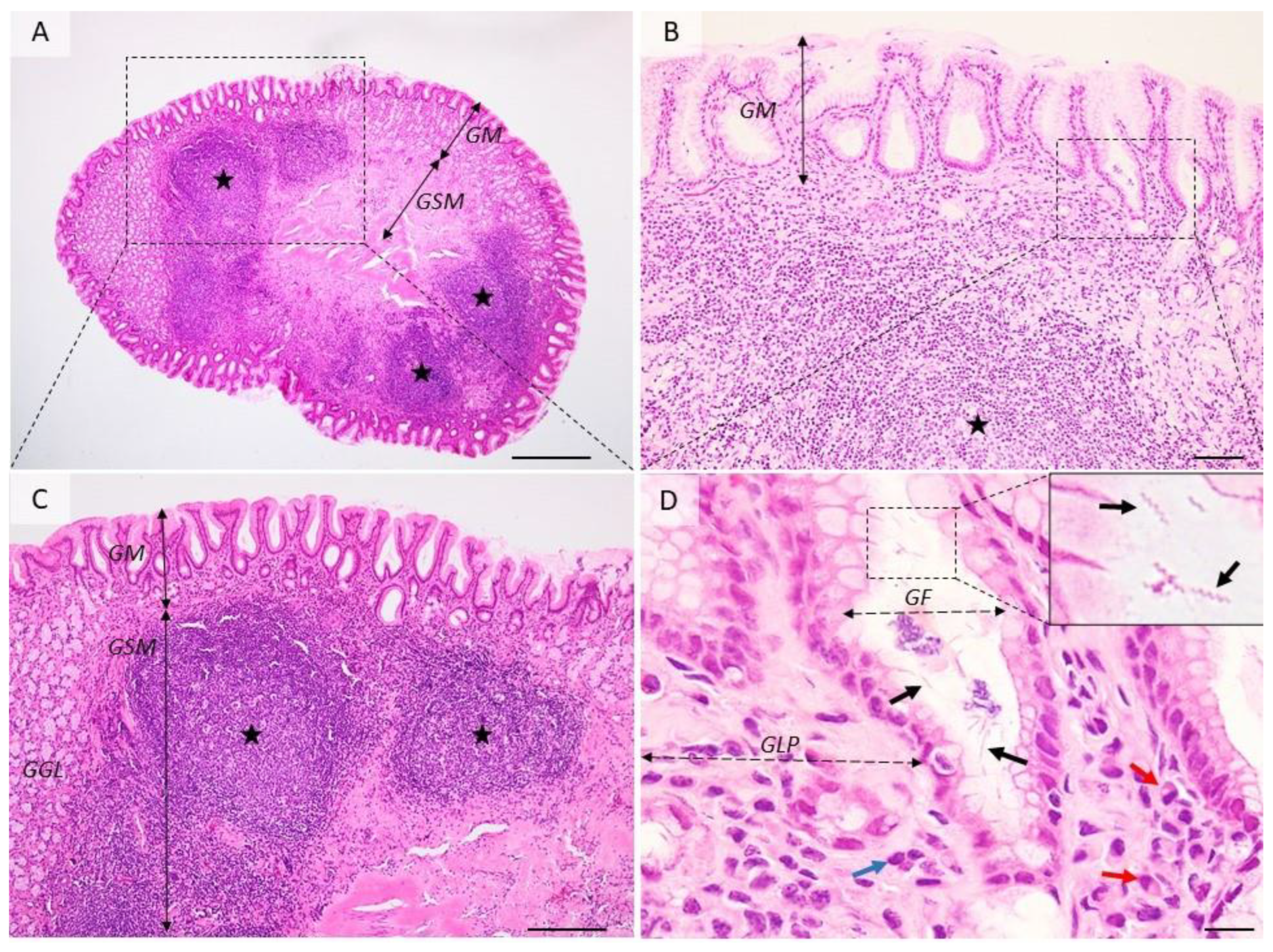
Figure 2. Histological aspects of follicular gastritis. (A–C) The lamina propria is multifocally distended and the gastric glands are displaced by many coalescing lymphoid follicles showing normal lymphocyte maturation. (D) Detailed demarcated area from (B). Within the gastric pits and superficial epithelium, there are many extracellular spiral-shaped bacteria measuring up to 7 µm. The lamina propria is infiltrated by many plasma cells (red arrow) and lymphocytes (blue arrow). GM = gastric mucosa; GSM = gastric submucosa; GGL = gastric glands; GF = gastric foveolar cells; GLP = gastric lamina propria; red arrows—plasma cells, blue arrow—fibroblasts, black arrows—Helicobacter spp.; black stars—lymphoid follicles. Dog, H&E stain, Ob × 4 (A), Ob × 20 (B), Ob × 10 (C), Ob × 100 (D); scale bar = 500 µm (A), 100 µm (B), 200 µm (C), 10 µm (D).
After the colonization of the gastric mucosa by H. pylori, the first disease produced by it is chronic active gastritis. Initially, chronic gastritis is superficial and limited to the antrum. As the infection persists, the lesions extend both in depth (atrophic gastritis) and on the surface of the gastric body (Figure 3). The persistence of injury to the gastric mucosa for a long time leads to the onset of hypo-/achlorhydria, maldigestion, malabsorption, and pernicious anemia [62]. The inflammatory process causes atrophy of mucosa and leads to intestinal metaplasia. The replacement of the gastric epithelium with the intestinal epithelium leads to the oncogenic risk increase and is proportional to the size of intestinal metaplasia areas. If the infection persists in time, in addition to the granulocyte infiltrate, lymphoid aggregates appear in the lamina propria of the mucosa, similar to lymphoid follicles (Peyer’s patches) in the small intestine (Figure 2). Gastric follicles become hyperplastic depending on the extension and duration of colonization of the stomach with H. pylori and are possibly at the origin of the development of a primary non-Hodgkin’s gastric lymphoma in the lymphoid tissue of the gastric mucosa [63].

Figure 3. Distribution of H. pylori in the gastric mucosa. (A) Diagram showing the main cell types present within the gastric mucosa. Helicobacter spp. are located extracellularly, within the mucus covering the superficial mucosa ((B), arrow), within the glandular lumen ((C), arrows), or, rarely, intracellularly, in specific vacuoles ((D), arrows). Dog, H&E stain, Ob × 100; scale bar = 10 µm. Created using BioRender.
Atrophic and metaplastic gastritis represents a chronic infection characterized histologically by a reduced number of epithelial cells and gastric glandular cells that evolve into foci or diffuse in gastric mucosa. It can be found as nodular gastritis, hemorrhagic gastritis, and hypertrophic gastritis [61].
Gastric ulcer is associated with antral gastritis in over 70% of cases. Erosions and gastric ulcers are the results of multifactorial action that alters the defensive barrier and the repair mechanisms of the gastric mucosa. All the factors that determine the increase in gastric acidity, the reduction of the recovery capacity of the epithelial cells predispose to the appearance of erosions and the formation of gastric ulcers [63]. The gastric mucosa is well-protected against bacterial infection, but H. pylori is well-adapted to this biological niche with unique characteristics, which allow it to enter the mucous layer, attach to epithelial cells, avoid the immune response, and, consequently, evoke persistent colonization and transmission. Mucosal colonization is due to a set of enzymes (urease, mucinase, peptidases, etc.) that provide it with pathogenicity, H. pylori being located under the mucous layer, between epithelial cells, and around the gastric crypts [64].
The emergence of gastric ulcers is due to two major pathogenic mechanisms: the direct one characterized by the decrease in mucosal resistance to aggressive factors where the inflammatory process initiated by Helicobacter toxins triggers acute gastritis that later becomes chronic and the indirect one characterized by stimulation of antral G cells, followed by increased gastric acid secretion and decreased secretion of somatostatin, all of which lead to an increased quantity of gastrin, especially postprandially [46].
2.2. Gastric Neoplasia Induced by H. pylori Infection
The gastric oncogenesis induced by H. pylori is complex and involves the sequential action of several pathogenicity factors (Table 1) and oncogenic mechanisms. There are several types of malignant gastric tumors induced by the chronic H. pylori infection, the most important in terms of both prevalence and aggressivity being gastric adenocarcinoma and gastric lymphoma.
Gastric adenocarcinoma. Gastric cancer is one of the most common types of tumors in the world. The fact that the major cause of gastric cancer is chronic H. pylori infection led to the classification of these bacteria in 1994 by the International Agency for Research on Cancer (IARC) under the World Health Organization as group I carcinogens, H. pylori being incriminated with certainty in the carcinogenesis of the gastric area. The recent studies of the experimental H. pylori infection of the Mongolian gerbil, an animal that is currently one of the model species for the study of Helicobacter-induced gastric tumors, have concluded that typical histopathological changes associated with this infection include chronic gastritis with subsequent metaplasia, intestinal and gastric mucosal atrophy. Most cases of gastric cancer develop into multifocal gastric atrophy, usually with extensive intestinal metaplasia, suggesting that metaplasia and gastric atrophy are precancerous lesions of the stomach [65][66]. H. pylori can produce tumor deviation of gastric cells in two ways: indirectly, by mutations of gastric epithelial cells as a collateral lesion of the inflammatory chain caused by the chronic presence of H. pylori, and directly, by the direct activity of the bacteria on the gastric epithelium or by the action of metabolites produced by the bacteria acting in the direction of alteration of the apoptosis/cell proliferation balance, impaired gene expression by action on transduction pathways, increased oxidative stress, alterations of intracellular toxic adhesion, direct action on epithelial cells, destruction of cellular DNA. Both bacterial proteins and cellular cytokines have a chemotactic role in inflammatory cells. Once activated, inflammatory cells produce chemical mediators represented by reactive oxygen species (ROS) while raising tissue levels of interleukin 8 (IL-8) [67].
Gastric lymphoma. The term “malignant lymphoma” suggests a more frequent malignant tumor of the lymph node tissue. In 1990, the association between H. pylori infection and gastric lymphoma was found; 92–97% of cases with gastric MALT lymphoma also showed lesions of chronic Helicobacter gastritis as well as the fact that antibiotic treatment to eradicate the infection may cause spontaneous remission of lymphoma in approximately 70% of patients, including treatment of gastric MALT lymphoma as a mandatory measure to eradicate H. pylori infection [68][69]. In humans, it has been established that in the case of Helicobacter infection, there is a 6.3% chance that dysplasia such as gastric MALT lymphoma will occur during life [66]. The mechanisms of action of H. pylori in the production of gastric MALT lymphoma are related to the proliferation of B lymphoma cells, proliferation stimulated by cytokines released by gastric T lymphocytes activated by the presence of H. pylori in the stomach [70]. MALT lymphoma occurs in normal lymphoid tissue but is preceded by the presence of chronic inflammation of lymphoid tissue, most commonly autoimmune. Paradoxically, although it does not normally have lymphoid tissue, the stomach is the most common site of extraganglionic lymphoma [71].
2.3. Extragastric Pathology Induced by H. pylori Infection
Although H. pylori infection is limited to the stomach, it triggers a significant systemic immune response in the patient. As a result, unfavorable effects of this reaction may lead to disease development in sites other than the gastrointestinal system.
Cardiovascular diseases. Lai et al. [72] have established that H. pylori infection significantly raises the risk of acute coronary syndrome. A meta-analysis of 26 studies including over 20,000 individuals performed by Liu et al. [70] found a significant link between H. pylori infection and the risk of myocardial infarction [73]. In patients with coronary artery disease (CAD), Lenzi et al. [74] investigated the overall prevalence of H. pylori and CagA-positive H. pylori infection, as well as the prevalence of other bacterial and viral causes of chronic infection, as well as the potential role of anti-heat shock protein 60 antibodies in increasing the risk of cardiovascular disease development [74]. H. pylori antibodies were found in the blood of patients with cardiovascular problems according to several seroepidemiologic studies. Atherosclerosis and chronic idiopathic thrombocytopenic purpura are two areas where H. pylori are likely to play a role, at least in a subset of individuals [75][76][77][78].
Hepatobiliary diseases. Hepatobiliary illnesses ranging from chronic cholecystitis and primary sclerosing cholangitis to gallbladder carcinoma and primary hepatic carcinomas have been linked to Helicobacter spp. that can invade the biliary tract. H. pylori have evolved adaptive methods to shield themselves from the stomach’s acidic environment. Helicobacter must have protection mechanisms against the negative effects of alkaline pH and bile acids if they are to survive in the biliary tract. Differential expression of virulence factors by different Helicobacter spp. is studied to see if it can help them survive in diverse habitats [79][80]. Bile acids are known to have inhibitory effects on the H. pylori. influence on adhesion and proliferation. Taurine-conjugated bile acids are better for H. pylori survival than glycine-conjugated bile acids. In an acidic environment, taurine-conjugated bile acids are more stable than glycine-conjugated bile acids, which can enhance the survivability of H. pylori. Although it is well-understood that bacteria play a role in the production of pigment stones, the particular mechanisms involved in stone formation are unknown. Interactions between Helicobacter species are also possible through the formation of hydrolyzing bile, nucleating proteins, chronic inflammation, and enzymes, microorganisms, or slime can act as a nidus for foreign bodies [81][82][83]. In a mouse model, Ki et al. [84] found that H. pylori increase hepatic fibrosis. Bacterial infection is thought to play a role in the production of cholesterol gallstones, and recent research has discovered the presence of Helicobacter spp. in the hepatobiliary system [85]. Helicobacter species DNA was discovered in the gallbladders of Chinese patients with gallstone illnesses, showing that Helicobacter infection may simply be a cofactor in the production of gallstones rather than a major contributor [86]. The presence of Helicobacter DNA was found to be associated with an increase in the proliferating cell nuclear antigen labeling index in the biliary epithelium, suggesting that Helicobacter spp. may play a role in the pathophysiology of hepatobiliary cancer by accelerating biliary cell kinetics [87].
References
- Ierardi, E.; Losurdo, G.; Mileti, A.; Paolillo, R.; Giorgio, F.; Principi, M.; Di Leo, A. The puzzle of coccoid forms of Helicobacter pylori: Beyond basic science. Antibiotics 2020, 9, 293.
- Thamphiwatana, S.; Gao, W.; Obonyo, M.; Zhang, L. In vivo treatment of Helicobacter pylori infection with liposomal linolenic acid reduces colonization and ameliorates inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 17600–17605.
- Wolle, K.; Malfertheiner, P. Treatment of Helicobacter pylori. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 315–324.
- Zullo, A.; Hassan, C.; Cristofari, F.; Andriani, A.; De Francesco, V.; Ierardi, E.; Tomao, S.; Stolte, M.; Morini, S.; Vaira, D. Effects of Helicobacter pylori eradication on early stage gastric mucosa–associated lymphoid tissue lymphoma. Clin. Gastroenterol. Hepatol. 2010, 8, 105–110.
- Franceschi, F.; Gasbarrini, A. Helicobacter pylori and extragastric diseases. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 325–334.
- Huang, W.-S.; Yang, T.-Y.; Shen, W.-C.; Lin, C.-L.; Lin, M.-C.; Kao, C.-H. Association between Helicobacter pylori infection and dementia. J. Clin. Neurosci. 2014, 21, 1355–1358.
- Shen, X.; Yang, H.; Wu, Y.; Zhang, D.; Jiang, H. Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 2017, 22, e12398.
- Álvarez-Arellano, L.; Maldonado-Bernal, C. Helicobacter pylori and neurological diseases: Married by the laws of inflammation. World J. Gastrointest. Pathophysiol. 2014, 5, 400.
- Blecker, U.; Renders, F.; Lanciers, S.; Vandenplas, Y. Syncopes leading to the diagnosis of a Helicobacter pylori positive chronic active haemorrhagic gastritis. Eur. J. Pediatr. 1991, 150, 560–561.
- Mendall, M.A.; Goggin, P.M.; Molineaux, N.; Levy, J.; Toosy, T.; Strachan, D.; Camm, A.J.; Northfield, T.C. Relation of Helicobacter pylori infection and coronary heart disease. Heart 1994, 71, 437–439.
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. 2018, 33, 33–43.
- Lai, L.H.; Sung, J.J. Helicobacter pylori and benign upper digestive disease. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 261–279.
- Robinson, K.; Argent, R.H.; Atherton, J.C. The inflammatory and immune response to Helicobacter pylori infection. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 237–259.
- Hsu, P.I.; Wu, D.-C.; Wu, J.-Y.; Graham, D.Y. Is there a benefit to extending the duration of Helicobacter pylori sequential therapy to 14 days? Helicobacter 2011, 16, 146.
- Zullo, A.; De Francesco, V.; Hassan, C.; Morini, S.; Vaira, D. The sequential therapy regimen for Helicobacter pylori eradication: A pooled-data analysis. Gut 2007, 56, 1353–1357.
- O’Morain, C.; Smith, S. Helicobacter pylori treatment failure: The rationale for alternative antibiotics. Digestion 2016, 93, 309–310.
- Pandya, H.B.; Agravat, H.H.; Patel, J.S.; Sodagar, N.R.K. Emerging antimicrobial resistance pattern of Helicobacter pylori in central Gujarat. Indian J. Med. Microbiol. 2014, 32, 408–413.
- Megraud, F.; Coenen, S.; Versporten, A.; Kist, M.; Lopez-Brea, M.; Hirschl, A.M.; Andersen, L.P.; Goossens, H.; Glupczynski, Y. Helicobacter pylori resistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 2013, 62, 34–42.
- Wang, B.; Lv, Z.F.; Wang, Y.H.; Wang, H.; Liu, X.-Q.; Xie, Y.; Zhou, X.-J. Standard triple therapy for Helicobacter pylori infection in China: A meta-analysis. World J. Gastroenterol. 2014, 20, 14973–14985.
- Liu, G.; Xu, X.; He, L.; Ding, Z.; Gu, Y.; Zhang, J.; Zhou, L. Primary antibiotic resistance of Helicobacter pylori isolated from Beijing children. Helicobacter 2011, 16, 356–362.
- Dang, B.N.; Graham, D.Y. Helicobacter pylori infection and antibiotic resistance: A WHO high priority? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 383–384.
- Kollef, M.H. Inadequate antimicrobial treatment: An important determinant of outcome for hospitalized patients. Clin. Infect.Dis. 2000, 31 (Suppl. S4), S131–S138.
- French, G.L. Clinical impact and relevance of antibiotic resistance. Adv. Drug Deliv.Rev. 2005, 57, 1514–1527.
- French, G.L. The continuing crisis in antibiotic resistance. Int. J. Antimicrob. Agents 2010, 36, S3–S7.
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of antibiotic resistance in Helicobacter pylori: A systematic review and meta-analysis in World Health Organization regions. Gastroenterology 2018, 155, 1372–1382.
- Megraud, F.; Bruyndonckx, R.; Coenen, S.; Wittkop, L.; Huang, T.-D.; Hoebeke, M.; Bénéjat, L.; Lehours, P.; Goossens, H.; Glupczynski, Y. Helicobacter pylori resistance to antibiotics in Europe in 2018 and its relationship to antibiotic consumption in the community. Gut 2021, 70, 1815–1822.
- Navarro-Rodriguez, T.; Silva, F.M.; Barbuti, R.C.; Mattar, R.; Moraes-Filho, J.P.; de Oliveira, M.N.; Bogsan, C.S.; Chinzon, D.; Eisig, J.N. Association of a Probiotic to a Helicobacter pylori Eradication Regimen Does Not Increase efficacy or decreases the adverse effects of the treatment: A prospective, randomized, double-blind, placebo-controlled study. BMC Gastroenterol. 2013, 13, 56.
- Akcam, M.; Koca, T.; Salman, H.; Karahan, N. The effects of probiotics on treatment of Helicobacter pylori eradication in children. Saudi Med. J. 2013, 36, 286.
- Hu, Y.; Zhu, Y.; Lu, N.-H. Novel and effective therapeutic regimens for Helicobacter pylori in an era of increasing antibiotic resistance. Front. Cell. Infect. Microbiol. 2017, 7, 168.
- Ko, S.W.; Kim, Y.; Chung, W.C.; Lee, S.J. Bismuth supplements as the first-line regimen for Helicobacter pylori eradication therapy: Systemic review and meta-analysis. Helicobacter 2019, 24, e12565.
- Kim, S.Y.; Choi, D.J.; Chung, J.-W. Antibiotic treatment for Helicobacter pylori: Is the end coming? World J. Gastrointest. Pharmacol. Ther. 2015, 6, 183.
- Amin, M.; Iqbal, M.S.; Hughes, R.W.; Khan, S.A.; Reynolds, P.A.; Enne, V.I.; Sajjad-ur-Rahman Mirza, A.S. Mechanochemical synthesis and in vitro anti-Helicobacter pylori and uresase inhibitory activities of novel zinc (II)–famotidine complex. J. Enzyme Inhib. Med. Chem. 2010, 25, 383–390.
- Bruggraber, S.F.; French, G.; Thompson, R.P.; Powell, J.J. Selective and effective bactericidal activity of the cobalt (II) cation against Helicobacter pylori. Helicobacter 2004, 9, 422–428.
- Vimbela, G.V.; Ngo, S.M.; Fraze, C.; Yang, L.; Stout, D.A. Antibacterial properties and toxicity from metallic nanomaterials. Int. J. Nanomedicine 2017, 12, 3941.
- Gopinath, V.; Priyadarshini, S.; MubarakAli, D.; Loke, M.F.; Thajuddin, N.; Alharbi, N.S.; Yadavalli, T.; Alagiri, M.; Vadivelu, J. Anti-Helicobacter pylori, cytotoxicity and catalytic activity of biosynthesized gold nanoparticles: Multifaceted application. Arab. J. Chem. 2019, 12, 33–40.
- Li, W.-R.; Xie, X.-B.; Shi, Q.-S.; Zeng, H.-Y.; You-Sheng, O.-Y.; Chen, Y.-B. Antibacterial activity and mechanism of silver nanoparticles on Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 85, 1115–1122.
- Geng, W.; Jiang, N.; Qing, G.-Y.; Liu, X.; Wang, L.; Busscher, H.J.; Tian, G.; Sun, T.; Montelongo, Y.; Janiak, C.; et al. Click reaction for reversible encapsulation of single yeast cells. ACS Nano 2019, 13, 14459–14467.
- Geng, W.; Wang, L.; Jiang, N.; Cao, J.; Xiao, Y.-X.; Wei, H.; Yetisen, A.K.; Yang, X.-Y.; Su, B.-L. Single cells in nanoshells for the functionalization of living cells. Nanoscale 2018, 10, 3112–3129.
- Tan, P.; Li, H.; Wang, J.; Gopinath, S.C. Silver nanoparticle in biosensor and bioimaging: Clinical perspectives. Biotechnol. Appl. Biochem. 2021, 68, 1236–1242.
- Bao, C.; Beziere, N.; del Pino, P.; Pelaz, B.; Estrada, G.; Tian, F.; Ntziachristos, V.; de la Fuente, J.M.; Cui, D. Gold nanoprisms as optoacoustic signal nanoamplifiers for in vivo bioimaging of gastrointestinal cancers. Small 2013, 9, 68–74.
- Chen, N.-T.; Tang, K.-C.; Chung, M.-F.; Cheng, S.-H.; Huang, C.-M.; Chu, C.-H.; Chou, P.-T.; Souris, J.S.; Chen, C.-T.; Mou, C.-Y.; et al. Enhanced plasmonic resonance energy transfer in mesoporous silica-encased gold nanorod for two-photon-activated photodynamic therapy. Theranostics 2014, 4, 798.
- Blaser, M. The role of Helicobacter pylori in gastritis and its progression to peptic ulcer disease. Aliment. Pharmacol. Ther. 1995, 9, 27–30.
- Audibert, C.; Burucoa, C.; Janvier, B.; Fauchere, J. Implication of the structure of the Helicobacter pylori cag pathogenicity island in induction of interleukin-8 secretion. Infect. Immun. 2001, 69, 1625–1629.
- Nilsson, C.; Sillén, A.; Eriksson, L.; Strand, M.-L.; Enroth, H.; Normark, S.; Falk, P.; Engstrand, L. Correlation between cag pathogenicity island composition and Helicobacter pylori-associated gastroduodenal disease. Infect. Immun. 2003, 71, 6573–6581.
- Hacker, J.; Blum-Oehler, G.; Mühldorfer, I.; Tschäpe, H. Pathogenicity islands of virulent bacteria: Structure, function and impact on microbial evolution. Mol. Microbiol. 1997, 23, 1089–1097.
- Queiroz, D.; Mendes, E.; Rocha, G.; Moura, S.; Resende, L.; Barbosa, A.; Coelho, L.; Passos, M.; Castro, L.; Oliveira, C. Effect of Helicobacter pylori eradication on antral gastrin-and somatostatin-immunoreactive cell density and gastrin and somatostatin concentrations. Scand. J. Gastroenterol. 1993, 28, 858–864.
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186.
- Kao, C.-Y.; Sheu, B.-S.; Wu, J.-J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23.
- Nomura, A.M.; Pérez-Pérez, G.I.; Lee, J.; Stemmermann, G.; Blaser, M.J. Relation between Helicobacter pylori cagA status and risk of peptic ulcer disease. Am. J. Epidemiol. 2002, 155, 1054–1059.
- Cover, T.L. The vacuolating cytotoxin of Helicobacter pylori. Mol. Microbiol. 1996, 20, 241–246.
- Park, S.M.; Park, J.; Kim, J.G.; Yoo, B.C. Relevance of vacA genotypes of Helicobacter pylori to cagA status and its clinical outcome. Korean J. Intern. Med. 2001, 16, 8.
- Miehlke, S.; Yu, J.; Schuppler, M.; Frings, C.; Kirsch, C.; Negraszus, N.; Morgner, A.; Stolte, M.; Ehninger, G.; Bayerdörffer, E. Helicobacter pylori vacA, iceA, and cagA status and pattern of gastritis in patients with malignant and benign gastroduodenal disease. Am. J. Gastroenterol. 2001, 96, 1008–1013.
- Peek, R.M., Jr.; Thompson, S.A.; Donahue, J.P.; Tham, K.T.; Atherton, J.C.; Blaser, M.J.; Miller, G.G. Adherence to gastric epithelial cells induces expression of a Helicobacter pylori gene, iceA, that is associated with clinical outcome. Proc. Assoc. Am. Physicians 1998, 110, 531–544.
- Ernst, P.B.; Gold, B.D. The disease spectrum of Helicobacter pylori: The immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 2000, 54, 615–640.
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653.
- McGee, D.J.; Mobley, H.L. Pathogenesis of Helicobacter pylori infection. Curr. Opin. Gastroenterol. 2000, 16, 24–31.
- Radosz-Komoniewska, H.; Bek, T.; Jóźwiak, J.; Martirosian, G. Pathogenicity of Helicobacter pylori infection. Clin. Microbiol. Infect. 2005, 11, 602–610.
- Chang, W.L.; Yeh, Y.C.; Sheu, B.S. The impacts of H. pylori virulence factors on the development of gastroduodenal diseases. J. Biomed. Sci. 2018, 25, 68.
- Kalali, B.; Mejías-Luque, R.; Javaheri, A.; Gerhard, M. H. pylori virulence factors: Influence on immune system and pathology. Mediat. Inflamm. 2014, 2014, 426309.
- Lee, S. Future candidates for indications of Helicobacter pylori eradication: Do the indications need to be revised? J. Gastroenterol. Hepatol. 2012, 27, 200–211.
- Lee, S.Y. Endoscopic gastritis: What does it mean? Dig. Dis.Sci. 2011, 56, 2209–2211.
- Leib, M. Therapy of GI Diseases: What’s New with Antiemetics, Antacids, and Probiotics. Available online: http://www.dvm360storage.com/cvc/proceedings/dc/Gastrointestinal%20Medicine/Leib/Leib,%20Michael_Therapy_GI_diseases_STYLED.pdf (accessed on 31 May 2021).
- Guilford, W.G.; Center, S.A.; Strombeck, D.R.; Williams, D.A.; Meyer, D.J. Strombeck’s Small Animal Gastroenterology; WB Saunders Co.: Philadelphia, PA, USA, 1990.
- Westblom, T.U.; Czinn, S.J.; Nedrud, J.G. Gastroduodenal Disease and Helicobacter pylori: Pathophysiology, Diagnosis and Treatment; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012.
- Blok, P.; Craanen, M.E.; Johan Offerhaus, G.; Dekker, W.; Kuipers, E.J.; Meuwissen, S.G.; Tytgat, G.N. Molecular alterations in early gastric carcinomas: No apparent correlation with Helicobacter pylori status. Am. J. Clin. Pathol. 1999, 111, 241–247.
- Villako, K.; Kekki, M.; Maaroos, H.-I.; Sipponen, P.; Uibo, R.; Tammur, R.; Tamm, A. Chronic gastritis: Progression of inflammation and atrophy in a six-year endoscopic follow-up of a random sample of 142 Estonian urban subjects. Scand. J. Gastroenterol. 1991, 26, 135–141.
- Shimada, T.; Watanabe, N.; Hiraishi, H.; Terano, A. Redox regulation of interleukin-8 expression in MKN28 cells. Dig. Dis. Sci. 1999, 44, 266–273.
- Montalban, C.; Santon, A.; Redondo, C.; García-Cosio, M.; Boixeda, D.; Vazquez-Sequeiros, E.; Norman, F.; de Argila, C.; Alvarez, I.; Abraira, V. Long-term persistence of molecular disease after histological remission in low-grade gastric MALT lymphoma treated with H. pylori eradication. Lack of association with translocation t (11; 18): A 10-year updated follow-up of a prospective study. Ann. Oncol. 2005, 16, 1539–1544.
- Ruskoné-Fourmestraux, A. Les lymphomes gastriques du MALT. Rev. Médecine Interne 2004, 25, 573–581.
- Hussell, T.; Isaacson, P.G.; Crabtree, J.; Dogan, A.; Spencer, J. Immunoglobulin specificity of low grade B cell gastrointestinal lymphoma of mucosa-associated lymphoid tissue (MALT) type. Am. J. Pathol. 1993, 142, 285.
- Wotherspoon, A. Gastric MALT lymphoma and Helicobacter pylori. Yale J. Biol. Med. 1996, 69, 61.
- Lai, C.-Y.; Yang, T.-Y.; Lin, C.-L.; Kao, C.-H. Helicobacter pylori infection and the risk of acute coronary syndrome: A nationwide retrospective cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 69–74.
- Liu, J.; Wang, F.; Shi, S. Helicobacter pylori infection increase the risk of myocardial infarction: A meta-analysis of 26 studies involving more than 20,000 participants. Helicobacter 2015, 20, 176–183.
- Lenzi, C.; Palazzuoli, A.; Giordano, N.; Alegente, G.; Gonnelli, C.; Campagna, M.S.; Santucci, A.; Sozzi, M.; Papakostas, P.; Rollo, F. H. pylori infection and systemic antibodies to CagA and heat shock protein 60 in patients with coronary heart disease. World J. Gastroenterol. WJG 2006, 12, 7815.
- Sawayama, Y.; Ariyama, I.; Hamada, M.; Otaguro, S.; Machi, T.; Taira, Y.; Hayashi, J. Association between chronic Helicobacter pylori infection and acute ischemic stroke: Fukuoka Harasanshin Atherosclerosis Trial (FHAT). Atherosclerosis 2005, 178, 303–309.
- Inaba, T.; Mizuno, M.; Take, S.; Suwaki, K.; Honda, T.; Kawai, K.; Fujita, M.; Tamura, T.; Yokota, K.; Oguma, K. Eradication of Helicobacter pylori increases platelet count in patients with idiopathic thrombocytopenic purpura in Japan. Eur. J. Clin. Investig. 2005, 35, 214–219.
- Ando, T.; Tsuzuki, T.; Mizuno, T.; Minami, M.; Ina, K.; Kusugami, K.; Takamatsu, J.; Adachi, K.; El-Omar, E.; Ohta, M. Characteristics of Helicobacter pylori-induced gastritis and the effect of H. pylori eradication in patients with chronic idiopathic thrombocytopenic purpura. Helicobacter 2004, 9, 443–452.
- Fujimura, K.; Kuwana, M.; Kurata, Y.; Imamura, M.; Harada, H.; Sakamaki, H.; Teramura, M.; Koda, K.; Nomura, S.; Sugihara, S. Is eradication therapy useful as the first line of treatment in Helicobacter pylori-positive idiopathic thrombocytopenic purpura? Analysis of 207 eradicated chronic ITP cases in Japan. Int. J. Hematol. 2005, 81, 162–168.
- Hynes, S.; McGuire, J.; Wadstrom, T. The effect of bile on protein expression by intestinal Helicobacters, determined using ProteinChip (r) Technology. Gut 2001, 49, A67.
- Leong, R.; Sung, J. Helicobacter species and hepatobiliary diseases. Aliment. Pharmacol. Ther. 2002, 16, 1037–1045.
- Harvey, P.; Upadhya, G.A.; Strasberg, S. Immunoglobulins as nucleating proteins in the gallbladder bile of patients with cholesterol gallstones. J. Biol. Chem. 1991, 266, 13996–14003.
- Swidsinski, A.; Lee, S.P. The role of bacteria in gallstone pathogenesis. Front. Biosci. 2001, 6, 93–103.
- Stewart, L.; Ponce, R.; Oesterk, A.L.; Griffiss, J.M.; Way, L.W. Pigment gallstone pathogenesis: Slime production by biliary bacteria is more important than beta-glucuronidase production. J. Gastrointest. Surg. 2000, 4, 547–553.
- Ki, M.-R.; Goo, M.-J.; Park, J.-K.; Hong, I.-H.; Ji, A.-R.; Han, S.-Y.; You, S.-Y.; Lee, E.-M.; Kim, A.-Y.; Park, S.-J. Helicobacter pylori accelerates hepatic fibrosis by sensitizing transforming growth factor-β 1-induced inflammatory signaling. Lab. Investig. 2010, 90, 1507–1516.
- Suzuki, H.; Franceschi, F.; Nishizawa, T.; Gasbarrini, A. Extragastric manifestations of Helicobacter pylori infection. Helicobacter 2011, 16, 65–69.
- Chen, W.; Li, D.; Cannan, R.; Stubbs, R. Common presence of Helicobacter DNA in the gallbladder of patients with gallstone diseases and controls. Dig. Liver Dis. 2003, 35, 237–243.
- Gasbarrini, A.; Carloni, E.; Gasbarrini, G.; Ménard, A. Helicobacter pylori and extragastric diseases–other Helicobacters. Helicobacter 2003, 8, 68–76.
More
Information
Subjects:
Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
27 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No