1. Introduction
It is true that medical treatment has brought a significant benefit to human health, but unmet medical needs still remain, particularly in the treatment of central nervous system (CNS) diseases
[1] and cancers. In particular, brain cancer drug development is, synergistically, an extraordinarily challenging task with respect to pharmacokinetics and pharmacodynamics. Drug delivery into the brain presents a serious impenetrable problem in CNS drug development, due to repulsion by the blood-brain barrier (BBB)
[2][3]. Most drugs cannot be transported from the systemic circulation to the brain across the BBB. The BBB is substantially composed of (i) a physical barrier based on hydrophobic lipid bilayer membrane, (ii) a physical barrier based on tight junctions between the capillary endothelial cells, (iii) a biological barrier based on efflux transporters such as multiple drug resistance 1 (MDR1) (P-glycoprotein), and (iv) a physical barrier based on a lining supported by pericytes and astrocytes. Accordingly, intravenously administered CNS drugs must cross the BBB to complete their activity in the target sites. Receptor-mediated transcytosis (RMT) represents one of the solutions to this impenetrability problem
[2]. Some receptors, such as the transferrin receptor (TfR) or insulin receptor (InsR), transport their corresponding ligands across the endothelium or the epithelium via RMT. So far, I have described several drug delivery methods across the cell membrane
[2][4][5][6][7][8][9] using RMT or carrier-mediated transport, based on rational drug design, particularly across the BBB
[2][9]. From such an investigative process, it has been revealed that pharmacokinetically effective drug delivery can be accomplished by rigorous design, regulated through physically and biologically systematic structures such as the BBB or RMT machinery system, based on the theory of structuralism proselytized by Lévi-Strauss. Compounds are divided into three categories, that is, low-molecular compounds (molecular weight (MW) < approximately 500), high-molecular compounds (MW > approximately 3000), and middle-molecular compounds (MW from approximately 500 to approximately 3000). In general, while hydrophobic low-molecular compounds penetrate the cell membrane through passive diffusion, hydrophilic low-molecular compounds penetrate it via carrier-mediated transport, using solute carrier (SLC) transporters. Water-soluble low-molecular nutrition, such as glucose and amino acids, are transported into the brain across the BBB by SLC transporters expressed at the BBB. High- or middle-molecular compounds penetrate the cell membrane through receptor-mediated endocytosis, lipid-raft mediated endocytosis, or macropinocytosis. High- or middle-molecular compounds cannot physically pass through the pores of SLC transporters due to their molecular size, although they can pass through transient disruption of tight junctions in the BBB. Bystander low-molecular compounds in the bloodstream are internalized into endosomes after spontaneous and receptor-mediated endocytosis, although they cannot induce endocytosis. Low-molecular compounds are subject to enzymatic metabolism and to excretion by kidneys into urine and by liver into bile. A drug delivery strategy can be naturally established, depending on which class of compounds in terms of size are used, in addition to their hydrophobicity and hydrophilicity (
Figure 1).
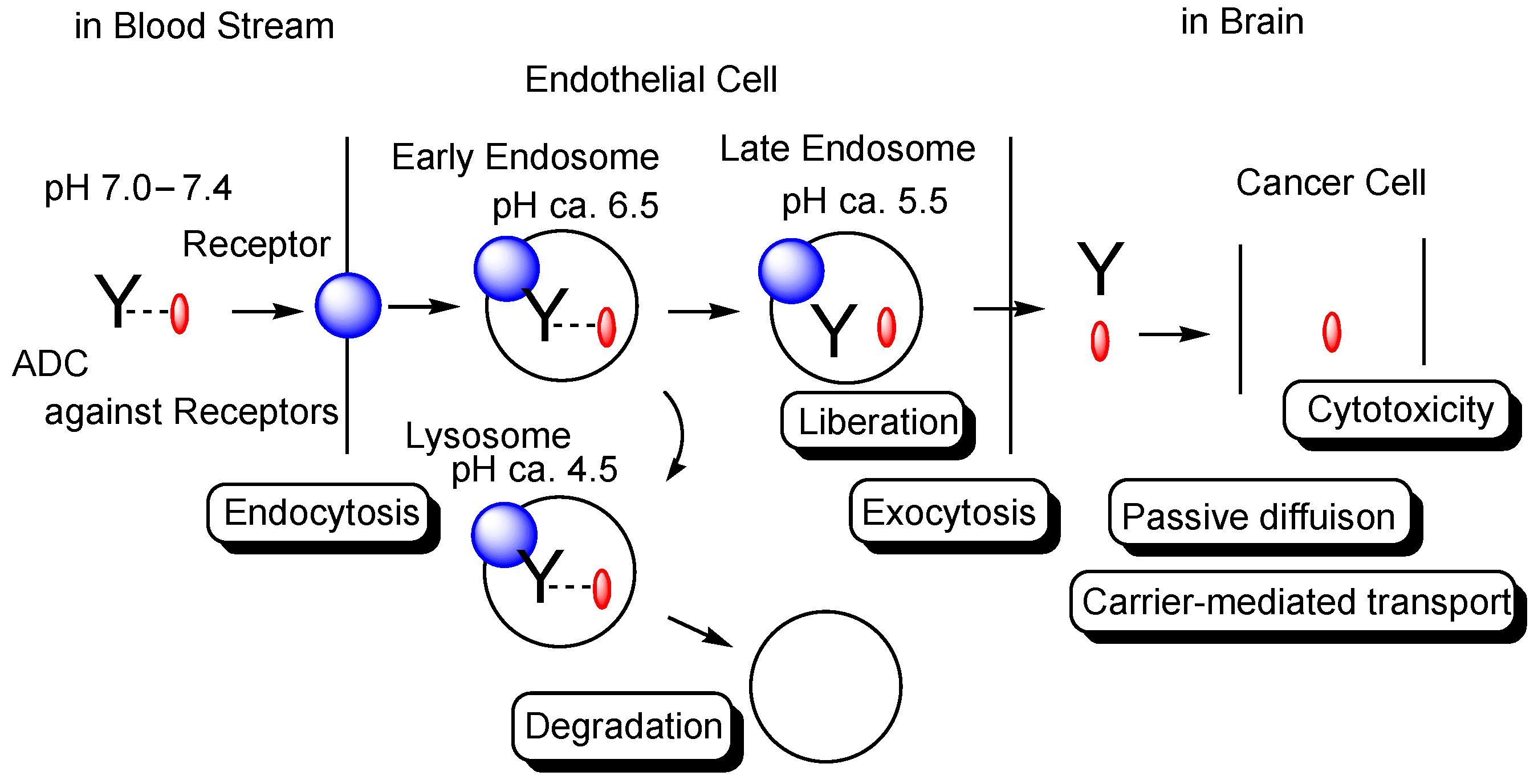
Figure 1. The pathway of intravenously administered antibody-drug conjugates (ADCs) against receptors such as the transferrin receptor (TfR), toward exhibiting brain cancer cell cytotoxicity through receptor-medicated transcytosis (RMT) in the secretory pathway. The mAb-TfR complex was liberated under weakly acidic conditions in the endosomes. Furthermore, linked drugs acting as a payload were also liberated via the cleavage of pH-sensitive cleavable linkers under weakly acidic conditions in the endosomes. Drugs released into the brain parenchyma can be transported into cancer cells and can show anti-cancer activity. Y represents a monoclonal antibody (mAb). The blue sphere indicates a receptor that mediates transcytosis in the capillary endothelial cells at the blood-brain barrier. The red ovals represent a drug that is tethered with a mAb through a suitable linker. The dotted line indicates a linker contained in an ADC. The solid line represents the membrane.
2. Brain Cancers and Their Chemotherapy
Cancer is a leading cause of death. Brain cancers are intracranial neoplasms that are either primary tumors or metastasizing tumors, occurring in approximately one hundred cases per one million people. They are broadly categorized into glioma, metastatic brain tumors, medulloblastoma, malignant lymphoma, germ cell tumors, meningioma, hypophyseal adenoma, and neurilemmoma. Treatments for brain cancers impose burdens on patients due to headaches, the consequences of surgical removal after craniotomy, and side effects from chemotherapy or radiation therapy. Thus, innovative brain cancer therapy is urgently needed to improve patients’ quality of life.
Glioma
[10], derived from glial cells, currently accounts for approximately one-third of brain cancers in Japan. The surgical removal of brain tumors, in some cases by labeling tumor cells with 5-aminolevulinic acid, is performed as a standard of care. Nonetheless, it is difficult to remove all tumor cells. Consequently, brain cancer chemotherapy, particularly for glioma, is generally conducted after tumor removal.
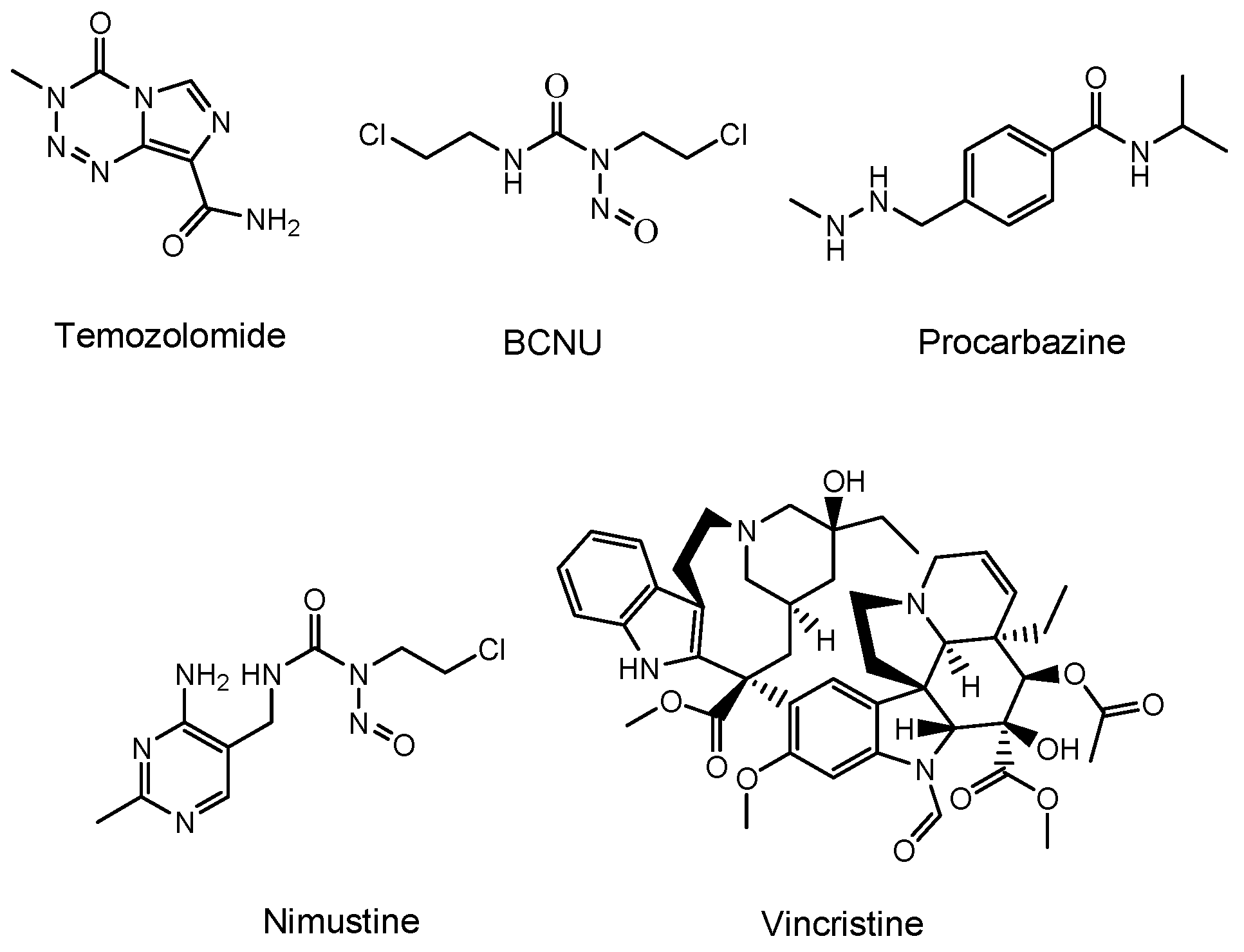
Temozolomide (an alkylating agent)
[11], bevacizumab (a monoclonal antibody (mAb) against vascular endothelial growth factor (VEGF) that is relevant in angiogenesis and is released from cancer cells)
[12], and a BCNU wafer (an alkylating slow-release agent)
[13] are often clinically prescribed for glioma (
Figure 2). Temozolomide is so hydrophobic that it penetrates the cell membrane via passive diffusion. However, MDR1 at the BBB captures hydrophobic low-molecular compounds that are just passing through the capillary endothelial cell membrane and excretes them to the systemic circulation. Thus, most of the temozolomide might be excreted by MDR1 at the BBB, without being distributed to the brain. Moreover, mAbs are so large and so hydrophilic that they cannot penetrates the cell membrane via passive diffusion. Thus, mAbs such as bevacizumab cannot penetrate the capillary endothelial cell membrane at the BBB, without being distributed to the brain. Furthermore, a BCNU wafer composed of polifeprosan 20, a degradable polymer, is left in the brain after tumor removal. However, the pharmacokinetics of polifeprosan 20 in the human brain remains unclarified, although, in the rat brain and rabbit brain, this was verified. Therefore, innovative drug delivery approaches should be developed, in order to carry out effective and harmless chemotherapy treatment.
Figure 2. Structures of low-molecular drugs that are used clinically for brain cancers.
3. Possibility and Implement of ADCs
Historically, biomedicines using mAbs have followed the genealogy from mAb drugs to ADCs, and further to bispecific mAbs. To deliver drugs to the brain across the BBB, the technologies of bispecific mAbs or bispecific ADCs through RMT are needed. In the future, trispecific mAbs will become a practical possibility. I will describe mAb biomedicines below, according to such a genealogy.
3.1. mAb Drugs
First of all, mAbs exhibit highly selective binding to the epitopes in their target antigen molecules. Ab drugs, as molecular targeted drugs, are representative biomedicines
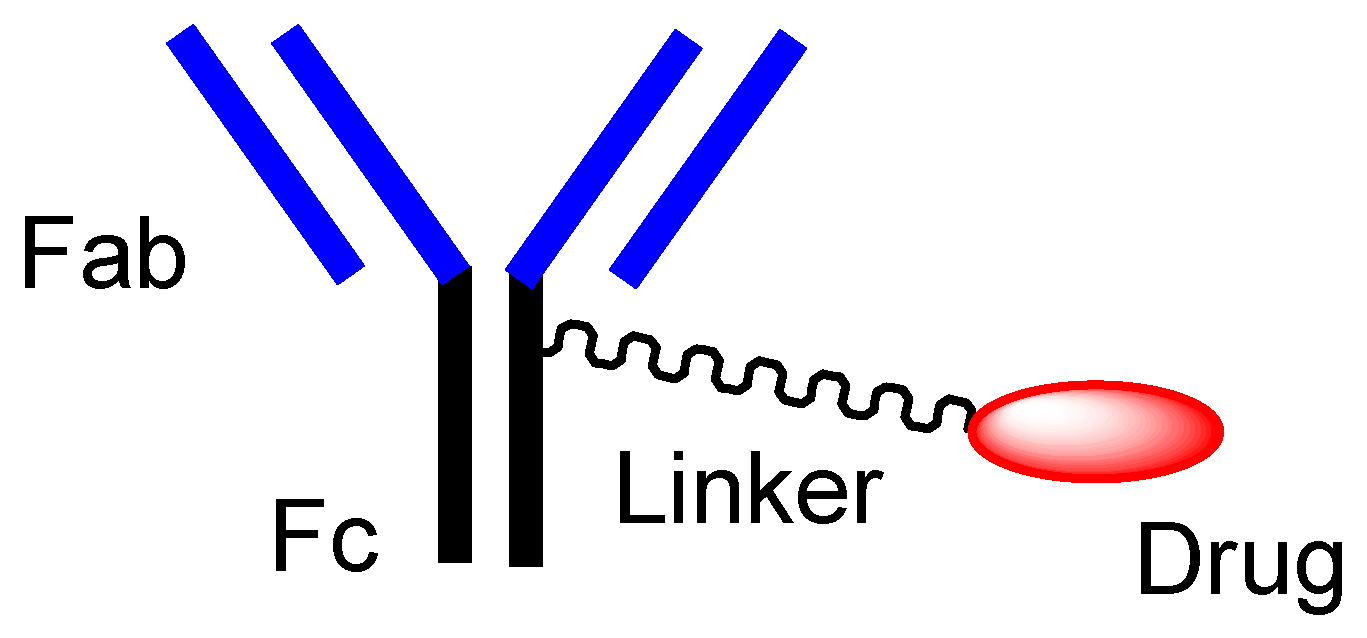
[14]. An Ab protein, such as immunoglobulin G (IgG), is structurally constituted of two heavy chains and two light chains and is enzymatically cleaved by papain into a fragment antigen-binding (Fab) region and a fragment crystallizable (Fc) region (
Figure 3). Japanese mAb technologies are excellent, and outstanding mAbs have been developed. Representative Ab production technologies include Ab manufacturing technology with high Ab-dependent cellular cytotoxicity (POTELLIGENT
®)
[15] by Kyowa Kirin (Tokyo, Japan), recycling Ab manufacturing technology (SMART-Ig
®)
[16] by Chugai (Tokyo, Japan), and bispecific Ab manufacturing technology (ART-Ig
®) by Chugai. Tocilizumab (Actemra
®)
[17], developed as an anti-IL-6 mAb by Chugai, was approved for rheumatoid arthritis treatment in 2010 by the US Food and Drug Administration (FDA). Nivolumab (Opdivo
®)
[18], developed as an anti-PD-L1 (programmed cell death 1) mAb by Ono (Osaka, Japan), was approved for metastatic lung squamous cell carcinoma treatment in 2014 by the FDA. Mogamulizumab (Poteligeo
®)
[19], developed as anti-CCR4 (CC chemokine receptor 4) mAb using POTELLIGENT
® by Kyowa Kirin, was approved for the treatment of relapsed or refractory mycosis fungoides and Sézary disease in 2018 by the FDA. Burosumab (Crysvita
®)
[20], developed as an anti-FGF23 (fibroblast growth factor 23) mAb by Kyowa Kirin, was approved for treating X-linked hypophosphatemic rickets in 2018 by the FDA.
Figure 3. The typical structure of antibody-drug conjugate (ADC).
3.2. Orthodox ADCs
ADCs comprise another area of development. The high selectivity of mAb enables specific distribution in terms of drug delivery. Thus, at present, ADCs, composed of an antibody as a vector and the drug as a payload via suitable linkers (
Figure 3), have attracted a great deal of attention
[6][21]. In the case of cancer therapies, antibody-cancer drug conjugates are thought to reduce off-target side effects, such as normal hematopoietic cell damage or hair loss, due to the specific interaction between epitopes as receptors that are specifically expressed on target cancer cells and mAbs as ligands, and due to the non-interaction between off-target molecules and linked cancer drugs, based on the bulkiness and large size of mAb. Such bulkiness might offer an escape from enzymatic drug degradation. If the Fc region of mAb is not occupied with payloads or linkers, the half-life of ADCs might elongate, based on salvation by the neonatal Fc receptor (FcRn) at the endothelium
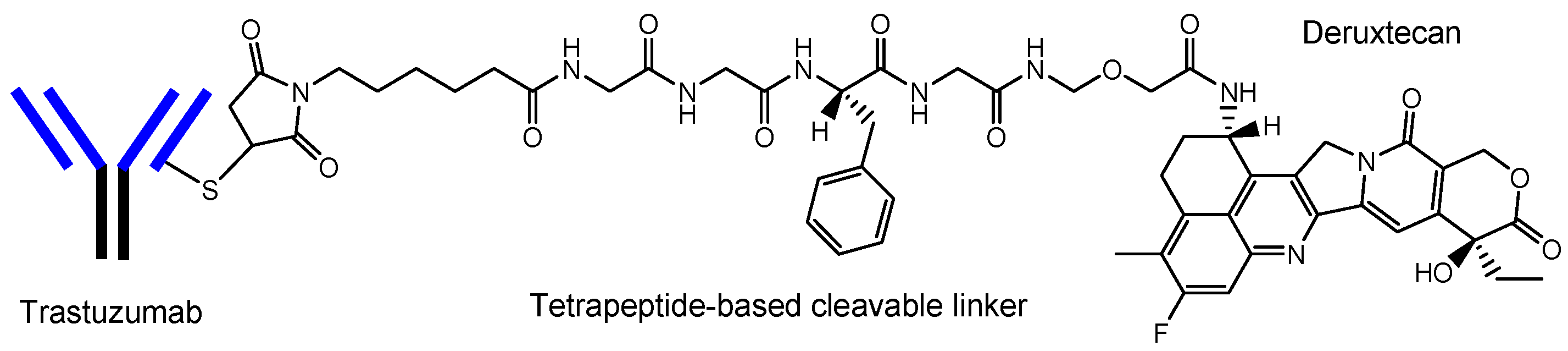
[8][22]. Actually, ADCs acting against the surface epitopes on cancer cells have been clinically utilized for solid cancers such as lung cancer, colon cancer, or breast cancer, and for blood cancers such as acute myelogenous leukemia. Trastuzumab deruxtecan (Enhertu
®) (
Figure 4)
[21], an anti-HER2 (human epidermal receptor 2) ADC developed by Daiichi Sankyo (Tokyo, Japan), was approved for HER2-positive breast cancer in 2019 by the FDA. Deruxtecan is a DNA topoisomerase I inhibitor. Trastuzumab deruxtecan was internalized into cancer cells via receptor-mediated endocytosis, using HER2
[23]. Furthermore, Daiichi Sankyo has developed (i) Dato-DXd as an anti-ROP2 ADC
[24], (ii) HER3-DXd as an anti-HER3 ADC
[25], (iii) DS-7300 as an anti-B7-H3 ADC
[26], (iv) DS-6000 as an anti-CDH6 ADC
[27], and (v) DS-3939 as an anti-TA-MUC1 ADC
[27], for solid cancers, respectively. Accordingly, ADCs might be ideal for use in brain cancer therapy, with respect to their effective distribution to the brain and low off-target side effects
[6]. However, surprisingly, ADCs for brain cancer have not been developed, probably due to poor distribution because of the BBB.
Figure 4. The structure of trastuzumab deruxtecan (Enhertu®), with a drug-to-antibody ratio (DAR) of 7.7.
3.3. Anti-Receptor ADCs That Cross the Endothelium via RMT
The enhanced permeation and retention (EPR) effect, discovered by Dr. Yasuhiro Matsumura and Dr. Hiroshi Maeda in 1986, spontaneously accumulates nanoparticles in cancer tissues, due to leaky blood vessels and hypervascularization
[6][28]. However, this cannot be expected in brain cancers due to the tight junctions at the BBB, although part of the membranes or tight junctions at the BBB might be disrupted as the result of pathological alteration. It is well known that RMT at the BBB, using TfR or InsR, is used for CNS drug delivery into the brain. Anti-TfR mAbs are endocytosed at the apical membrane of the capillary endothelial cells after ligand-receptor binding, are then liberated from TfRs through acidification in the endosomes, and are, finally, released into the brain via exocytosis, based on the fusion between the endosomes and the basolateral membrane. Moreover, mAbs acting as ligands showed higher selectivity than the cell-penetrating peptides (CPPs) such as TAT (YGRKKRRQRRR) and penetratin (RQIKIWFQNRRMKWKK). CPPs are positively charged short peptides, with 5–30 amino acids, that cross the cell membrane through receptor-mediated endocytosis or by direct translocation
[5]. Although TAT, as a ligand, induced receptor-mediated endocytosis using the negatively charged heparan sulfate proteoglycans (HSPGs) on the cell surface, HSPGs in the form of receptors are ubiquitously expressed on many types of cells. Interestingly, J-Brain Cargo
® (
Figure 5)
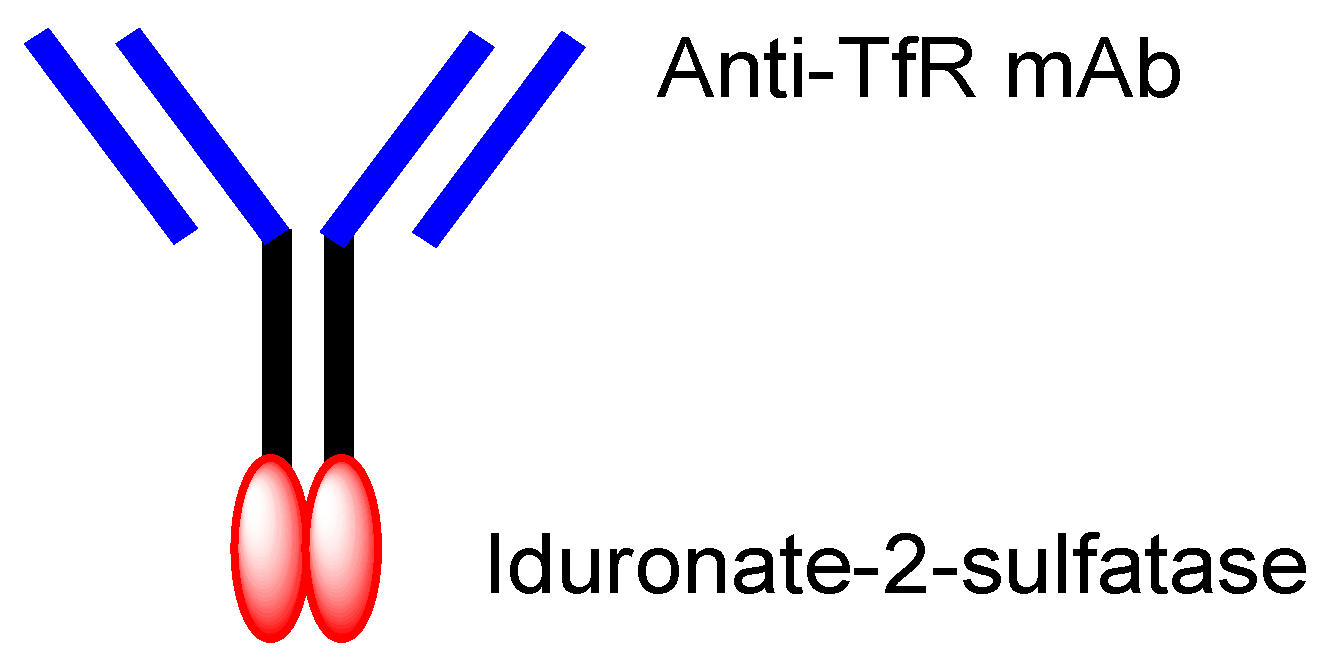
[2][29], established by JCR Pharmaceuticals (Ashiya, Japan), is a system to deliver drugs into the brain across the BBB via RMT, using ADCs composed of anti-TfR mAb and drugs. This system is used by the drug idursulfase beta (
Figure 5), approved in Japan on 23 March 2021 for the treatment of Hunter syndrome
[30]. Takeda (Tokyo and Osaka, Japan) is going to sell idursulfase beta worldwide, together with JCR Pharmaceuticals. The concept idea of idursulfase beta, based on J-Brain Cargo
®, can be technologically adapted to cancer therapy by replacing the payloads. Once ADCs are transported into the brain parenchyma via RMT, they or their liberated payloads can stay there without being excreted out of the brain by the BBB. This feature might help to reduce off-target side effects outside the brain.
Figure 5. The structure of idursulfase beta, composed of anti-TfR (transferrin receptor) mAb (monoclonal antibody) and iduronate-2-sulfatase.
3.4. Anti-Receptor ADCs including Bispecific ADCs
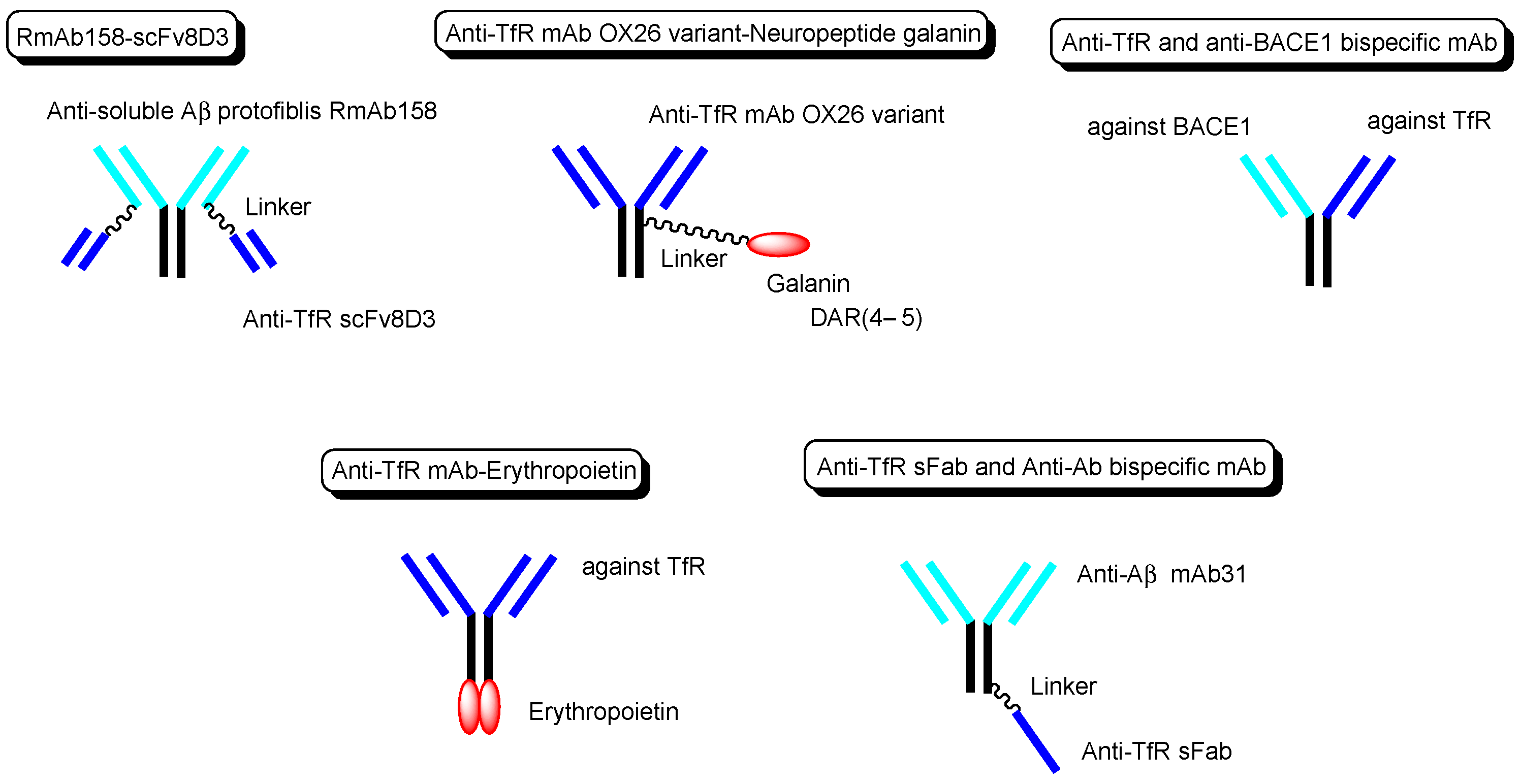
Other technical cases of drug delivery across the BBB via RMT using anti-TfR mAbs as a vector are shown here, including anti-receptor and anti-target molecule bispecific technologies. (i) Bispecific RmAb158-scFv8D3 (
Figure 6) is a conjugate of scFv8D3 with two single-chain (sc) variable fragments of anti-TfR Ab 8D3 and RmAb158 against soluble amyloid β (Aβ) protofibrils, through the linker at each
C-terminus of scFv8D3, and two light chains of RmAb158. Actually, [
125I]-RmAb158-scFv8D3 was transported 80-fold higher in the brain than [
125I]-RmAb158 in an in vivo assay with mice euthanized 2 h after intravenous injection
[31]. (ii) The conjugate of the anti-TfR mAb OX26 variant and neuropeptide galanin (YPSKPDNPGEDAPAEDMARYYSALRHYINLITRQRY), with a drug-to-antibody ratio (DAR) of 4–5 at cysteine residues on the Fc region of the OX26 variant through a maleimide group (
Figure 6), inhibited thermal hyperalgesia in an in vivo assay intravenously administered in a rat model, compared to the conjugate of galanin and control mAb NiP228
[32]. (iii) Intravenously administered anti-TfR and anti-BACE1 (β-amyloid cleaving enzyme-1) bispecific mAb (
Figure 6) reduced brain Aβ levels in an in vivo assay using mice.
[33]. (iv) Anti-TfR mAb, when fused to erythropoietin (
Figure 6), significantly lowered cortical and hippocampal Aβ peptide levels, decreased hippocampal synaptic loss, decreased cortical microglial activation, and improved spatial memory in an APP/PS1 mouse model of Alzheimer’s disease (AD)
[34]. (v) The monovalent conjugate of one scFab fragment of an anti-TfR mAb and anti-Aβ mAb31, linked at one
C-terminal end of the heavy chain of anti-Aβ mAb (
Figure 6), was transported into the brain across the BBB via RMT and exhibited 55-fold higher Aβ plaque binding than the parent mAb31 after intravenous administration, in an in vivo assay using PS2APP transgenic mice
[35]. Interestingly, the bivalent conjugate of two scFab fragments of an anti-TfR mAb and anti-Aβ mAb31, linked at one
C-terminal end of the heavy chain of anti-Aβ mAb31 with one scFab fragment for each, bound simultaneously to two TfRs and subsequently degraded as the generated complex in lysosomes after receptor-mediated endocytosis. Incidentally, bispecific RmAb158-scFv8D3 bound one TfR due to structural restriction and was endocytosed, although it had two scFv8D3
[31].
Figure 6. The structures of anti-TfR mAbs, conjugated to active cargos.
Nonetheless, the orthodox type of anti-TfR ADCs, with linked low-molecular brain cancer drugs (Figure 3), is not likely to be developed. Therefore, well-designed brain cancer drug delivery into the brain via RMT, using anti-receptor mAbs as a vector, has great possibilities.
4. Activity Expression of ADCs
Activity expression by the payloads of ADCs assumes two scenarios, such as (i) the liberation of payloads after linker cleavage, or (ii) no structural alteration of ADCs, including bispecific mAbs or ADC with relatively bare payloads, such as idursulfase beta. If low-molecular cancer drugs were conjugated as a payload, they would be cut off from ADCs due to the bulkiness of mAb, shown in the curious case of AMG-595, below. Several linker cleavage systems have been developed
[6]: (a) pH-sensitive cleavable linkers, such as hydrazone, which can be cleaved in an acidic endosome, (b) reductively cleavable linkers, such as the disulfide bond, (c) enzymatically cleavable linkers, such as valine-citrulline dipeptide, (d) self-immolative linkers, such as the para-aminobenzyloxycarbonyl group, and (e) other mechanistically cleavable linkers, such as a photocleavable component.
The extracellular physiological pH is between 7.0 and 7.4. The pH in endosomes gradually reduces from the early endosome (pH of approximately 6.5) to the late endosome (pH of approximately 5.5) and, finally, lysosome (pH of approximately 4.5) by vacuolar H
+-ATPase proton pumps. Thus, ideally, the linker of the anti-TfR mAb-cancer drug conjugate should be cleaved in endosomal acidification. In fact, pH-sensitive cleavable linkers were developed. At the same time, anti-TfR mAbs should be liberated from TfRs under weakly acidic conditions, without being degraded in lysosomes in the degradation pathway. When anti-TfR mAbs bind TfR too tightly, the resulting unliberated complexes are degraded into pieces in lysosomes by the lysosomal enzymes. Actually, the affinity of anti-TfR mAbs to TfR can be tuned so as to be separated as endosomal acidification. Eventually, free cancer drugs are released into the brain after exocytosis in the secretory pathway (
Figure 1)
[2]. The released drugs move within the brain parenchyma, due to restricted distribution by the BBB and, subsequently, elicit their activity in target sites on the surface of cancer cells or in cancer cells after internalization via passive diffusion or carrier-mediated transportation. Intriguingly, the FcRn-IgG complex is formed in the early endosomes because FcRn binds the Fc region of IgG under weakly acidic conditions (pH < 6.5)
[8]. The FcRn-IgG complex is exocytosed to the systemic circulation in the secretory pathway and is liberated under extracellular physiological pH. This system by FcRn at the endothelium results in the long half-life of ADCs, due to their salvation from lysosomal degradation. Therefore, drug designs should be carried out in the context of the involved physically and biologically systematic structures.
On the other hand, reductively cleavable linkers, enzymatically cleavable linkers, and other mechanistically cleavable linkers, such as a photocleavable component, should be cleaved in the brain parenchyma. Orthodox ADCs that had already crossed the BBB via RMT would probably not enter the brain cancer cells via RMT again; therefore, they might release their payloads in the brain parenchyma.
 +1 credit
+1 credit