Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Florian Schmid | -- | 2821 | 2022-07-15 12:38:42 | | | |
| 2 | Sirius Huang | Meta information modification | 2821 | 2022-07-20 02:46:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Schmid, F.; Chao, C.; Däbritz, J. Pathophysiological Concepts of Pulmonary Manifestation of Pediatric IBD. Encyclopedia. Available online: https://encyclopedia.pub/entry/25279 (accessed on 24 July 2026).
Schmid F, Chao C, Däbritz J. Pathophysiological Concepts of Pulmonary Manifestation of Pediatric IBD. Encyclopedia. Available at: https://encyclopedia.pub/entry/25279. Accessed July 24, 2026.
Schmid, Florian, Cho-Ming Chao, Jan Däbritz. "Pathophysiological Concepts of Pulmonary Manifestation of Pediatric IBD" Encyclopedia, https://encyclopedia.pub/entry/25279 (accessed July 24, 2026).
Schmid, F., Chao, C., & Däbritz, J. (2022, July 19). Pathophysiological Concepts of Pulmonary Manifestation of Pediatric IBD. In Encyclopedia. https://encyclopedia.pub/entry/25279
Schmid, Florian, et al. "Pathophysiological Concepts of Pulmonary Manifestation of Pediatric IBD." Encyclopedia. Web. 19 July, 2022.
Copy Citation
Pulmonary manifestation (PM) of inflammatory bowel disease (IBD) in children is a rare condition. The exact pathogenesis is still unclear, but several explanatory concepts were postulated and several case reports in children were published. Different pathophysiological concepts were identified, including the shared embryological origin, “miss-homing” of intestinal based neutrophils and T lymphocytes, inflammatory triggering via certain molecules (tripeptide proline-glycine-proline, interleukin 25), genetic factors and alterations in the microbiome.
children
inflammation
immunity
gut–lung axis
airways
pulmonary function tests
pulmonary manifestation
1. Introduction
Inflammatory bowel disease (IBD) has shown an increasing prevalence in recent decades worldwide [1]. In 25% of patients, IBD manifestation occurs before the age of 18 [1]. Under the umbrella term of IBD, three different disease entities are subsumed: Crohn’s disease (CD), ulcerative colitis (UC) and inflammatory bowel disease—unspecified [2]. While CD potentially occurs in any part of the digestive tract from mouth to anus and shows an incontiguous, transmural inflammation, UC usually is an ascending, continuous inflammation beginning at the rectal mucosa. The currently accepted pathophysiological idea comprises a dysregulation of the enteric immune system in genetically predisposed individuals, which results in an inadequate response to commensal enteric bacteria [3]. However, IBD is not only understood as an intestinal, but also as a systemic disease, which can affect different organs, called extraintestinal manifestations (EIM) of IBD [4][5]. In the adult IBD population, approximately 30% develop EIM and a quarter of them even before the onset of IBD [6]. If one other organ beside the gut is affected, the likelihood of suffering from further EIM is significantly increased [7]. The prevalence of EIM in the pediatric population was reported to be from 17% to 28% after 15 years of diagnosis [8][9][10]. While 6% of pediatric patients develop EIM before the diagnosis of IBD, one fourth show EIM in the first 2 to 3 years after the diagnosis [10]. Almost ninety percent of EIM will occur in the first year of IBD course [9]. The most common EIM are arthritis, skin lesions (i.e., erythema nodosa and pyoderma gangrenosum), aphthous stomatitis, uveitis and hepatobiliary diseases (i.e., primary sclerosing cholangitis and hepatitis) [10][11][12]. The pulmonary system as a location of EIM is a rare condition (<1%), interstitial pneumonitis being seen more in UC patients and granulomatous disease being more associated with CD [13][14][15]. While in adults pulmonary manifestation (PM) is generally more detected in UC patients, most of the reported cases in children and adolescents are associated with CD [16]. Forty percent of adult IBD patients show respiratory alterations, which are frequently subclinical and might not be clinically relevant [12]. PM in pediatric IBD patients has rarely been described, mostly by case reports [17]. In a large cohort study of >1600 children with IBD, none showed PM [10]. However, it has to be noted that this number might be underrated, due to the fact that the majority of pediatric IBD patients with PM are asymptomatic. Furthermore, no regular screening for pulmonary disease has been established to date in the pediatric IBD population [18][19].
Since the first report of a patient with IBD and pulmonary symptoms in 1976 by Kraft el al., several pathophysiological concepts for PM have been proposed [20].
2. Pathophysiological Concepts
2.1. Shared Embryological Origin
Most of the described links between pulmonary and intestinal tissue are based on the shared embryological origin in the foregut portion of the endoderm, leading to the possibility of a cross-mucosal interaction between the two organs that compose the human mucosal immune system [21][22]. Although much of this gut–lung interaction is not finally understood, both organs show a similar architecture, being composed of an extensive mucosal surface (with goblet cells and submucosal glands), containing lymphoid tissue (innate and adaptive immunity) and being covered by mucus, commensal bacteria and other antigens [16][23][24][25]. The gut and lung share the ability of a mucosal defense mechanism [17]. For instance, the dendritic cells of the lung are able to upregulate the expression of α4β7-integrine, which results in T lymphocyte (Tc) migration to the gut and therefore a direct communication pathway between the intestine and the lung [26]. Furthermore, due to the similar structure of these two mucosal organs, Papanikolaou et al. hypothesized that circulating autoantibodies, such as perinuclear anti-neutrophil cytoplasmic antibodies (pANCA) or anti-saccharomyces cerevisiae antibodies (ASCA), might lead to similar effects of inflammation in the lung as in the gut tissue [16].
2.2. Innate and Adaptive Immunity
In IBD, the homeostasis of the intestinal mucosal immune system is unbalanced, which results in a dysregulated response to luminal, commensal bacteria and other antigens [3]. The first step towards this dysregulation consists of a leaky intestinal mucus and interepithelial cell connection, which facilitates antigens to translocate through the intestinal epithelium [27][28]. In a second step, dendritic cells recognize these antigens as pathogens and induce a T-cell-mediated immune response [29]. Furthermore, activated macrophages induce the expression of leucocyte adhesion molecules at the endothelial blood vessels via the secretion of cytokines (interleukin 1 (IL-1) and tumor necrosis factor alpha (TNF-α)) [30]. This neutrophilic-mediated inflammation plays a key role in IBD development [5]. Neutrophilic cells (Nc) are generated from myeloid precursor cells in the bone marrow, induced by granulocyte colony stimulating factor and IL-17A and then released to the blood stream [31]. Passing by inflamed tissue, Nc become tethered via P-selectin glycoprotein ligand 1 to selectins (P-selectin and E-selectin) expressed by endothelium cells (EC) and consequently migrate into the inflamed tissue (termed margination and diapedesis) [32]. It was shown that the amount of margination and diapedesis of Nc in the pulmonary vasculature during systemic inflammation is augmented, which might be one reason for the trapping of neutrophils in the lung during active IBD [33]. Furthermore, in contrast to other organs, the neutrophilic emigration in the lung vasculature mainly takes place in smaller capillaries and not in post-capillary venules with a larger diameter. Therefore, the pooling time of Nc in the lung is prolonged [34][35]. Moreover, it was shown that pulmonary permeability is increased in patients with CD, facilitating the emigration of Nc [36]. Yipp et al. were able to demonstrate via confocal pulmonary intravital microscopy that, in bacteriemia, pulmonary Nc detect bacterial endotoxins (lipopolysaccharide) via Toll-like receptor 4 (TLR4) binding, resulting in the cluster of differentiation (CD)11b-dependent crawling of Nc. This finally leads to a quicker response to bacterial pathogens compared to the TLR4-myeloid-differentiation primary response gene 88 (Myd88) activation of macrophages. This activation hence triggers, via nuclear factor kappa B (NF-κB), the subsequent expression of TNF-α, chemokines and other proinflammatory molecules, apart from activation of EC to produce selectins for Nc migration [37]. In IBD patients, Nc become initially primed in the gut mucosa during episodes of disease flare. A subset of the these gut-primed Nc are able to relocate from the abluminal to the luminal membrane (reverse transendothelial migration (rTEM)) [38]. Furthermore, the inflamed gut tissue leads to an expression of IL-6, TNF-α, interferon-γ and vascular endothelial growth factor (VEGF) [39][40][41]. Elevated systemic levels of these factors result in an upregulation of ligands for extravasation of Nc and an increased vascular permeability in lung tissue [5][42]. Therefore, the primed (rTEM) Nc recirculate to the bloodstream and translocate into the respiratory endothelium [5]. In vivo studies reveale, that junctional adhesion molecule C is suppressed in inflamed, hypoxic tissue, leading to an increased rTEM of Nc. In addition, primed Nc change their shape and though become deformable, which results in a prolonged transit time through the pulmonary vasculature [38][43]. Interestingly, the healthy lung serves as an organ of de-priming Nc to stabilize the neutrophile homeostasis. In the injured lung, though, this function is compromised, leading to more primed Nc passing the pulmonary blood pool and therefore more Nc transmigrating via the arterial blood stream into the pulmonary mucosa. This finally leads to an exaggerated neutrophilic-mediated inflammation [44]. Aydin et al. found significantly increased VEGF and TNF-α concentration in lung tissue during experimental colitis in rats [45]. TNF-α not only leads to neutrophile accumulation and enhanced expression of adhesion molecules, but also to the formation of granuloma, resulting in the granulomatous manifestation of IBD in the lung [46]. Overall, it can be stated that the close relationship of the mucosal immune systems of the gut and lung via primed (rTEM) Nc might contribute to PM.
Tc represent a second cell type that exhibits a potential mechanism to promote pulmonary inflammation and subsequent tissue damage in the lung. In healthy organs, Tc tend to migrate to the same tissue, where they were first exposed to their specific antigen [47]. Unlike the specific attraction of gut memory Tc via the interaction of α4β7–integrin on Tc and mucosal addressin cell adhesion molecule 1 on intestinal EC, the migration of these Tc to the pulmonary system is based on an unspecific mechanism [48]. The non-tissue-specific binding of L-selectin/CD62L on Tc to peripheral lymph node addressin on EC and/or α4β1-integrin on Tc to vascular cell adhesion molecule 1 on EC results in the migration of lymphocytes to the bronchus-associated lymphatic tissue (BALT) [49]. It was shown that pulmonary Tc express further non-tissue-specific receptors termed C-C chemokine receptor 3 (CCR3) and C-X-C chemokine receptor 5 (CXCR5), which are both increasingly expressed in the Tc of inflamed intestinal tissue [50][51]. Therefore, memory Tc, which experienced their first contact to a specific antigen in the inflamed intestinal mucosa, carry a high number of CCR3 and CXCR5 and, as a result, can translocate to the BALT. This called “miss-homing” explains the presence of gut memory Tc in lung tissue, but it requires an initiation via a gut-specific stimulus to start the inflammatory process. The necessary antigens might derive from a breakdown of the intestinal mucosal barrier during IBD [52].
IL-25 is expressed by respiratory and intestinal epithelial cells and was shown to play an important role in the development of two inflammatory diseases—IBD and bronchial asthma [53][54][55][56]. Experiments in mice revealed that the neutralization of IL-25 results in a decreased inflammation of the colonic mucosa and diminished levels of pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α [57]. However, in contrast, IL-25 levels are decreased in the colonic biopsies of CD patients and furthermore pro-inflammatory IL-13 is suppressed by the binding of IL-25 to a specific receptor at CD14+ monocyte-like cells [58]. Therefore, in IBD patients, IL-25 implies a chimeric pathogenic and protective function [59]. While in UC patients inflammation is trigged via T helper cells 2 (Th2), in CD, the expression of Th1-derived cytokines leads to intestinal inflammation [3]. IL-25 is mainly expressed by Th2 [60]. Therefore, it is not surprising that the pro-inflammatory effect of IL-25 is more evident in UC. IL-25 also plays a role in pulmonary inflammation upon allergen provocation [61].
2.3. Further Molecular Concepts
The tripeptide proline-glycine-proline (PGP), formed from collagen by two enzymes (matrix metalloproteinase [MMP] and prolyl endopeptidase [PE]), has a chemotactic function for Nc in the intestine by binding to the chemokine receptor CXCR2 on neutrophils [62]. The expression of MMP and PE is elevated in intestinal inflammation. Therefore, more Nc are being attracted, which are able to express MMP and PE themselves [62]. This subsequently results in a neutrophil-mediated intestinal collagen proteolysis and therefore a vicious circle of neutrophilic inflammation [62]. This was shown earlier for inflammatory diseases of the lung, such as chronic obstructive pulmonary disease and asthma, and though it might be one cause of PM of IBD, to date no studies regarding the concentration of PGP in sputum of IBD patients have been published [63].
The already mentioned damage to the epithelial barrier seen in IBD leads to significant bacteremia [64]. This in turn results in neutrophil accumulation in the lung tissue, which is positively associated with levels of IL-1β and C-C-ligand 2 (CCL2), which play a role in pulmonary inflammatory diseases (i.e., bronchiectasis and chronic bronchitis) [65]. Liu et al. discovered, in murine models, that this neutrophil recruitment in the lung is triggered by the expression of platelet-activating factor receptor (PAFR), which then results in the overexpression of the inflammasome via nucleotide-binding oligomerization domain-containing protein (NOD)-like receptor family pyrin-domain-containing 3 (NLRP3) [66]. Therefore, not the actual bacterial colonization, but the activation of lung-hosted inflammasome might play a role in the development of PM [66]. Furthermore, the correlation between an enhanced expression of PAFR and diminished levels of L-selectin on Nc contributes to the trapping of Nc on the endothelial surface and hence the further activation of the inflammation processes [67].
Liu et al. were further able to demonstrate that inflammatory damage to the mucosa and microvascular endothelial changes, such as increased angiogenesis, adhesion molecule expression for lymphocytes (i.e., intercellular adhesion molecule 1), leukocyte extravasation and decreased endothelial barrier function, are similar in the intestinal and pulmonary mucosa during UC [68]. Thromboxane B2 is increasingly expressed in the lung and colonic tissue of murine UC models, which augments leukocyte recruitment and results in mucosal injury via the activation and aggregation of platelets, VEGF-A, which leads to angiogenesis, and finally inducible nitric oxide synthase (iNOS) enhancing leukocyte adhesion [68].
2.4. Microbiome
It seems inevitable that the intestinal microbiome plays an important role in the disease development of IBD [69]. Microbial exposure during early life not only decreases the probability of developing IBD later in childhood or adulthood, but also of other chronic inflammatory diseases occurring in pediatric patients, such as asthma [70][71]. Studying sensitization to food allergens, Stefka et al. discovered that fewer Clostridia spp. result in a dysregulated homeostasis of the host–commensal relationship via reduced IL-22 expression. This finally leads to the alteration of the epithelial junction integrity and the secretion of epithelial antimicrobial peptides [72]. Olszak et al. were able to demonstrate that invariant natural killer T cells accumulate in the lamina propria of the murine gut as well of the lung tissue after contact with commensal microbes early in life. This accumulation is associated with an elevated expression of chemokine ligand CXCL3, which then leads to more lymphatic migration and consequently to IBD or asthma [73].
2.5. Genetic Factors
The concept of IBD as an “organ barrier disease” might underpin a further connection between two organs with a large mucosal surface—the intestine and the lung. Around 40% of CD patients show disease-associated variants of the caspase recruitment domain-containing protein 15 (CARD15) gene, coding for the intracellular NOD2 receptor [74]. These mutations cannot be stated as alone standing factors for their contribution to IBD development, as the genetic susceptibility involvement is thought to be around 20% [75]. The NOD2 receptor, which is not only present in macrophages and dendritic cells, but also in intestinal epithelial cells, is part of a family of pathogen recognition receptors (PRR), recognizing muramyl dipeptide (MDP) as part of the bacterial cell wall [76][77]. The MDP-NOD2-mediated pathway leads via NF-κB activation and translocation to the nucleus to an enhanced expression of α-defensins. In 2004, Wehkamp et al. first discovered that the mutation of NOD2 in patients with CD results in a diminished expression of these α-defensins leading to a breakdown of the mucosal barrier [78]. Therefore, it can be hypothesized that in IBD patients with PM the NOD2 mutation might not only lead to altered inflammation in the gut, but also on the lung surface, especially because NOD2 mutations were also described in the pathogenesis of another inflammatory pulmonary disease—chronic obstructive pulmonary disease (COPD) [79]. Mutations in the autophagy-related 16-like 1 (ATG16L1) gene lead to a diminished function of Paneth cells, a defective antigen presentation and proinflammatory cytokine secretion in IBD patients [80]. ATG16L1 function in PM was not studied, so a concluding assessment, whether the mutation is another link between the intestine and the lung, cannot be made. While specific expressions of humane leucocyte antigen (HLA) are associated with the development of EIM, such as HLA-A2 and HLA-DR1 in CD and HLA-B27, HLA-B8/DR3 and HLA-B58 in UC, no linkage to HLA-types were found for PM [16][81][82].
Figure 1 summarizes the current knowledge about potential cellular and molecular mechanisms involved in the development of PM in patients with IBD.
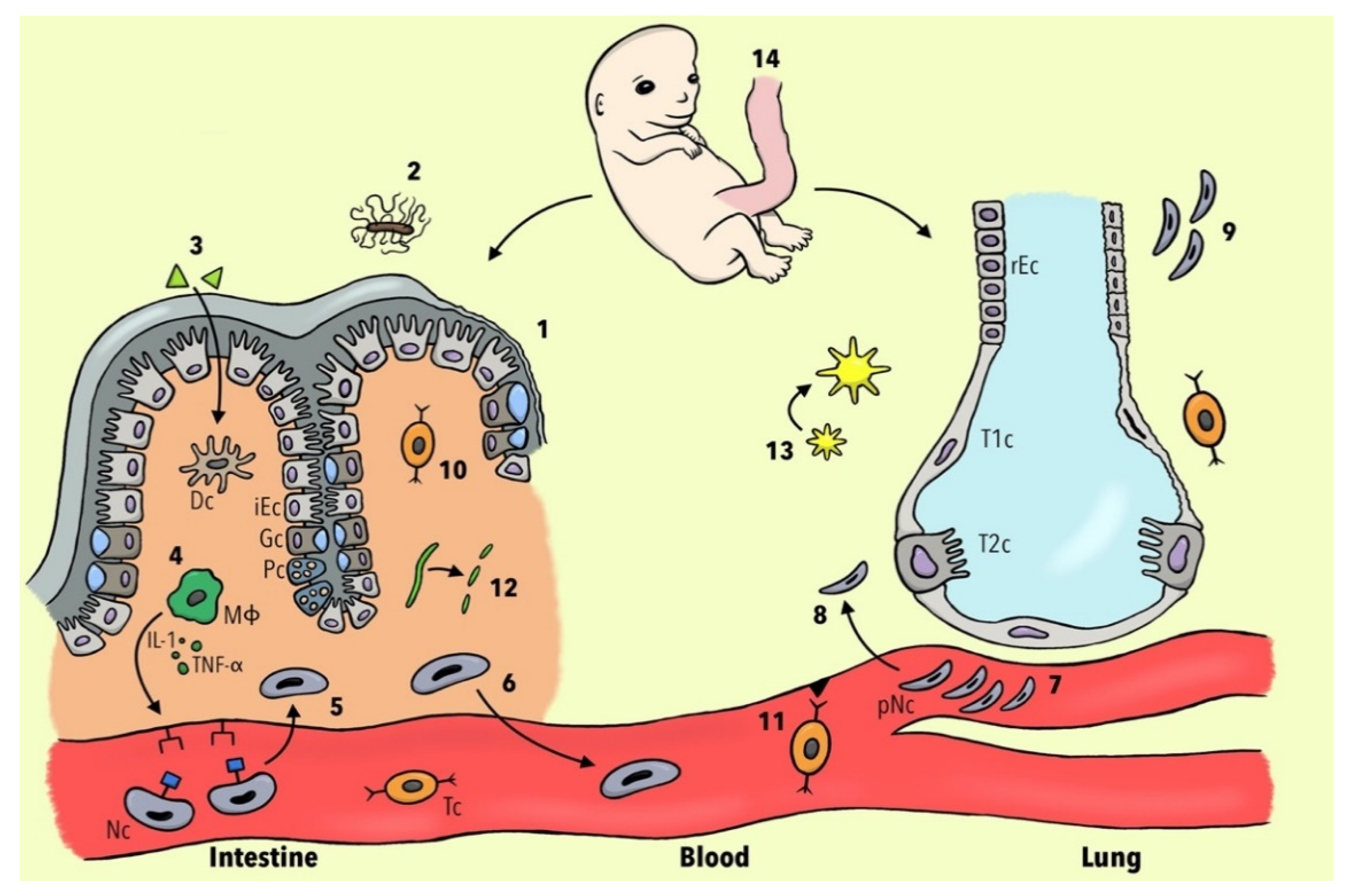
Figure 1. Different pathophysiological concepts of pulmonary manifestations in IBD patients. (1) Mutation of the CARD-15 gene leads to a diminished expression of α-defensins and consequently to a breakdown of the mucosal barrier. (2) Less Clostridia spp. result in dysregulation of host–commensal relationship and, via the reduction of IL-22, to the alteration of epithelial junction integrity. (3) Luminal antigens penetrate through the leaky intestinal mucosa and activate dendritic cells (DC). (4) Activated macrophages (ΜΦ) lead, via IL-1 and TNF-α, to the expression of neutrophils (Nc) adhesion molecules. (5) Nc undergo margination and diapedesis to the intestinal mucosa. (6) Nc become primed and a subset re-migrates to the bloodstream. (7) Nc change their shape and become deformable, resulting in a prolonged transit time in pulmonary capillaries. (8) JAM-C becomes downregulated and therefore the translocation of Nc to respiratory epithelium is facilitated. (9) The injured lung is not able to de-prime Nc leading to more neutrophilic inflammation. (10) Inflamed intestinal tissue leads to the upregulation of non-tissue-specific T-cell receptors CCR3 and CXCR5. (11) Diapedesis of gut-memory T cells (Tc)) via non-specific endothelial receptors to lung tissue is increased. (12) Collagen becomes degraded to tripeptide proline-glycine-proline, which plays a role in chemotaxis of Nc. (13) Increased bacterial burden leads to the expression of platelet-activating factor receptor and subsequently to the overexpression of the inflammasome. (14) Gut and lung tissues share the same embryological origin in the foregut portion of the endoderm. Abbreviations: iEC, intestinal epithelial cell; rEC, respiratory epithelial cell; Gc, goblet cell; Pc, Paneth cell; T1c, Type 1 pneumocytes; T2c, Type 2 pneumocytes; IL-1, interleukin 1; TNF-α, tumor necrosis factor alpha; pNC, primed neutrophils.
References
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van Den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of Pediatric Inflammatory Bowel Disease: A Systematic Review of International Trends. Inflamm. Bowel Dis. 2011, 17, 423–439.
- Bousvaros, A.; Antonioli, D.; Colletti, R.; Dubinsky, M.; Glickman, J.; Gold, B.; Griffiths, A.; Jevon, G.; Higuchi, L.; Hyams, J.; et al. Differentiating Ulcerative Colitis from Crohn Disease in Children and Young Adults: Report of a Working Group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn’s and Colitis Foundation of America. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 653–674.
- Baumgart, D.C.; Carding, S.R. Inflammatory Bowel Disease: Cause and Immunobiology. Lancet 2007, 369, 1627–1640.
- Hedin, C.R.H.; Vavricka, S.R.; Stagg, A.J.; Schoepfer, A.; Raine, T.; Puig, L.; Pleyer, U.; Navarini, A.; Van Der Meulen-De Jong, A.E.; Maul, J.; et al. The Pathogenesis of Extraintestinal Manifestations: Implications for IBD Research, Diagnosis, and Therapy. J. Crohn’s Colitis 2019, 13, 541–554.
- Mateer, S.W.; Maltby, S.; Marks, E.; Foster, P.S.; Horvat, J.C.; Hansbro, P.M.; Keely, S. Potential Mechanisms Regulating Pulmonary Pathology in Inflammatory Bowel Disease. J. Leukoc. Biol. 2015, 98, 727–737.
- Vavricka, S.R.; Rogler, G.; Gantenbein, C.; Spoerri, M.; Vavricka, M.P.; Navarini, A.A.; French, L.E.; Safroneeva, E.; Fournier, N.; Straumann, A.; et al. Chronological Order of Appearance of Extraintestinal Manifestations Relative to the Time of IBD Diagnosis in the Swiss Inflammatory Bowel Disease Cohort. Inflamm. Bowel Dis. 2015, 21, 1794–1800.
- Veloso, F.T.; Carvalho, J.; Magro, F. Immune-Related Systemic Manifestations of Inflammatory Bowel Disease: A Prospective Study of 792 Patients. J. Clin. Gastroenterol. 1996, 23, 29–34.
- Greuter, T.; Bertoldo, F.; Rechner, R.; Straumann, A.; Biedermann, L.; Zeitz, J.; Misselwitz, B.; Scharl, M.; Rogler, G.; Safroneeva, E.; et al. Extraintestinal Manifestations of Pediatric Inflammatory Bowel Disease: Prevalence, Presentation, and Anti-TNF Treatment. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 200–206.
- Dotson, J.L.; Hyams, J.S.; Markowitz, J.; Leleiko, N.S.; MacK, D.R.; Evans, J.S.; Pfefferkorn, M.D.; Griffiths, A.M.; Otley, A.R.; Bousvaros, A.; et al. Extraintestinal Manifestations of Pediatric Inflammatory Bowel Disease and Their Relation to Disease Type and Severity. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 140–145.
- Jose, F.A.; Garnett, E.A.; Vittinghoff, E.; Ferry, G.D.; Winter, H.S.; Baldassana, R.N.; Kirschner, B.S.; Cohen, S.A.; Gold, B.D.; Abramson, O.; et al. Development of Extraintestinal Manifestations in Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2009, 15, 63–68.
- Al-Binali, A.M.; Scott, B.; Al-Garni, A.; Montgomery, M.; Robertson, M. Granulomatous Pulmonary Disease in a Child: An Unusual Presentation of Crohn’s Disease. Pediatr. Pulmonol. 2003, 36, 76–80.
- Cozzi, D.; Moroni, C.; Addeo, G.; Danti, G.; Lanzetta, M.M.; Cavigli, E.; Falchini, M.; Marra, F.; Piccolo, C.L.; Brunese, L.; et al. Radiological Patterns of Lung Involvement in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 2018, 5697846.
- Eliadou, E.; Moleiro, J.; Ribaldone, D.G.; Astegiano, M.; Rothfuss, K.; Taxonera, C.; Ghalim, F.; Carbonnel, F.; Verstockt, B.; Festa, S.; et al. Interstitial and Granulomatous Lung Disease in Inflammatory Bowel Disease Patients. J. Crohn’s Colitis 2020, 14, 480–489.
- Inoue, K.; Kusunoki, T.; Fujii, T. Organizing Pneumonia as an Extraintestinal Manifestation of Crohn’s Disease in a Child. Pediatr. Pulmonol. 2017, 52, e64–e66.
- Rogers, B.H.G.; Clark, L.M.; Kirsner, J.B. The Epidemiologic and Demographic Characteristics of Inflammatory Bowel Disease: An Analysis of a Computerized File of 1400 Patients. J. Chronic Dis. 1971, 24, 743–773.
- Papanikolaou, I.; Kagouridis, M.; Papiris, S. Patterns of Airway Involvement in Inflammatory Bowel Diseases. World J. Gastrointest. Pathophysiol. 2014, 5, 560.
- Vadlamudi, N.B.; Navaneethan, U.; Thame, K.A.; Kelly, D.R.; Dimmitt, R.A.; Harris, W.T. Crohn’s Disease with Pulmonary Manifestations in Children: 2 Case Reports and Review of the Literature. J. Crohns Colitis 2013, 7, e85–e92.
- Taylor, M.A.; Zhou, H.; Dansie, D.M.; Short, S. Pulmonary Manifestations of Inflammatory Bowel Disease in Children. Cureus 2020, 12, e11369.
- Barfield, E.; Deshmukh, F.; Slighton, E.; Lentine, J.; Lu, Y.; Ma, X.; Christos, P.J.; Sockolow, R.E.; Loughlin, G.; Pillai, S. Pulmonary Manifestations in Adolescents With Inflammatory Bowel Disease. Clin. Pediatr. 2020, 59, 573–579.
- Kraft, S.C.; Earle, R.H.; Roesler, M.; Esterly, J.R. Unexplained Bronchopulmonary Disease With Inflammatory Bowel Disease. Arch. Intern. Med. 1976, 136, 454–459.
- Zhang, M.; Zhou, Y.; Li, H.; Peng, Y.; Qiu, P.; Shi, X.; Pan, X.; Liu, J. COVID-19: Gastrointestinal Symptoms from the View of Gut-Lung Axis. Eur. J. Gastroenterol. Hepatol. 2021, 33, 610–612.
- Shu, W.; Lu, M.M.; Zhang, Y.; Tucker, P.W.; Zhou, D.; Morrisey, E.E. Foxp2 and Foxp1 Cooperatively Regulate Lung and Esophagus Development. Development 2007, 134, 1991–2000.
- Keely, S.; Talley, N.J.; Hansbro, P.M. Pulmonary-Intestinal Cross-Talk in Mucosal Inflammatory Disease. Mucosal Immunol. 2012, 5, 7–18.
- Mestecky, J. The Common Mucosal Immune System and Current Strategies for Induction of Immune Responses in External Secretions. J. Clin. Immunol. 1987, 7, 265–276.
- Gut, G.; Sivan, Y. Respiratory Involvement in Children with Inflammatory Bowel Disease. Pediatr. Allergy Immunol. Pulmonol. 2011, 24, 197–206.
- Ruane, D.; Brane, L.; Reis, B.S.; Cheong, C.; Poles, J.; Do, Y.; Zhu, H.; Velinzon, K.; Choi, J.H.; Studt, N.; et al. Lung Dendritic Cells Induce Migration of Protective T Cells to the Gastrointestinal Tract. J. Exp. Med. 2013, 210, 1871–1888.
- Söderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzén, L.E.; Lindmark, T.; Wirén, M.; Tagesson, C.; Sjódahl, R. Augmented Increase in Tight Junction Permeability by Luminal Stimuli in the Non-Inflamed Ileum of Crohn’s Disease. Gut 2002, 50, 307–313.
- Johansson, M.E.V.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria Penetrate the Normally Impenetrable Inner Colon Mucus Layer in Both Murine Colitis Models and Patients with Ulcerative Colitis. Gut 2014, 63, 281–291.
- Bernardo, D.; Chaparro, M.; Gisbert, J.P. Human Intestinal Dendritic Cells in Inflammatory Bowel Diseases. Mol. Nutr. Food Res. 2018, 62, e1700931.
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621.
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689.
- Haslett, C.; Worthen, G.S.; Giclas, P.C.; Morrison, D.C.; Henson, J.E.; Henson, P.M. The Pulmonary Vascular Sequestration of Neutrophils in Endotoxemia Is Initiated by an Effect of Endotoxin on the Neutrophil in the Rabbit. Am. Rev. Respir. Dis. 1987, 136, 9–18.
- Lien, D.C.; Wagner, W.W.; Capen, R.L.; Haslett, C.; Hanson, W.L.; Hofmeister, S.E.; Henson, P.M.; Worthen, G.S. Physiological Neutrophil Sequestration in the Lung: Visual Evidence for Localization in Capillaries. J. Appl. Physiol. 1987, 62, 1236–1243.
- Hogg, J.C.; Coxson, H.O.; Brumwell, M.L.; Beyers, N.; Doerschuk, C.M.; MacNee, W.; Wiggs, B.R. Erythrocyte and Polymorphonuclear Cell Transit Time and Concentration in Human Pulmonary Capillaries. J. Appl. Physiol. 1994, 77, 1795–1800.
- Adenis, A.; Colombel, J.F.; Lecouffe, P.; Wallaert, B.; Hecquet, B.; Marchandise, X.; Cortot, A. Increased Pulmonary and Intestinal Permeability in Crohn’s Disease. Gut 1992, 33, 678–682.
- Yipp, B.G.; Kim, J.H.; Lima, R.; Zbytnuik, L.D.; Petri, B.; Swanlund, N.; Ho, M.; Szeto, V.G.; Tak, T.; Koenderman, L.; et al. The Lung Is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci. Immunol. 2017, 2, eaam8929.
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The Junctional Adhesion Molecule JAM-C Regulates Polarized Transendothelial Migration of Neutrophils in Vivo. Nat. Immunol. 2011, 12, 761–769.
- Funderburg, N.T.; Stubblefield Park, S.R.; Sung, H.C.; Hardy, G.; Clagett, B.; Ignatz-Hoover, J.; Harding, C.V.; Fu, P.; Katz, J.A.; Lederman, M.M.; et al. Circulating CD4(+) and CD8(+) T Cells Are Activated in Inflammatory Bowel Disease and Are Associated with Plasma Markers of Inflammation. Immunology 2013, 140, 87–97.
- Scaldaferri, F.; Vetrano, S.; Sans, M.; Arena, V.; Straface, G.; Stigliano, E.; Repici, A.; Sturm, A.; Malesci, A.; Panes, J.; et al. VEGF-A Links Angiogenesis and Inflammation in Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2009, 136, 585–595.e5.
- Raddatz, D.; Bockemühl, M.; Ramadori, G. Quantitative Measurement of Cytokine MRNA in Inflammatory Bowel Disease: Relation to Clinical and Endoscopic Activity and Outcome. Eur. J. Gastroenterol. Hepatol. 2005, 17, 547–557.
- Le Cras, T.D.; Spitzmiller, R.E.; Albertine, K.H.; Greenberg, J.M.; Whitsett, J.A.; Akeson, A.L. VEGF Causes Pulmonary Hemorrhage, Hemosiderosis, and Air Space Enlargement in Neonatal Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L134–L142.
- Downey, G.P.; Doherty, D.E.; Schwab, B.; Elson, E.L.; Henson, P.M.; Worthen, G.S. Retention of Leukocytes in Capillaries: Role of Cell Size and Deformability. J. Appl. Physiol. 1990, 69, 1767–1778.
- Nahum, A.; Chamberlin, W.; Sznajder, J.I.; Hegarty, M. Differential Activation of Mixed Venous and Arterial Neutrophils in Patients with Sepsis Syndrome and Acute Lung Injury. Am. Rev. Respir. Dis. 1991, 143, 1083–1087.
- Aydin, B.; Songur, Y.; Songur, N.; Aksu, O.; Senol, A.; Metin Ciris, I.; Sutcu, R. Investigation of Pulmonary Involvement in Inflammatory Bowel Disease in an Experimental Model of Colitis. Korean J. Intern. Med. 2016, 31, 853–859.
- Vilcek, J.; Lee, T.H. Tumor Necrosis Factor. New Insights into the Molecular Mechanisms of Its Multiple Actions. J. Biol. Chem. 1991, 266, 7313–7316.
- Campbell, D.J.; Butcher, E.C. Brief Definitive Report Rapid Acquisition of Tissue-Specific Homing Phenotypes by CD4 T Cells Activated in Cutaneous or Mucosal Lymphoid Tissues. J. Exp. Med. 2002, 195, 135–141.
- von Andrian, U.H.; Mackay, C.R. T-Cell Function and Migration. Two Sides of the Same Coin. N. Engl. J. Med. 2000, 343, 1020–1034.
- Xu, B.; Wagner, N.; Pham, L.N.; Magno, V.; Shan, Z.; Butcher, E.C.; Michie, S.A. Lymphocyte Homing to Bronchus-Associated Lymphoid Tissue (BALT) Is Mediated by L-Selectin/PNAd, Alpha4beta1 Integrin/VCAM-1, and LFA-1 Adhesion Pathways. J. Exp. Med. 2003, 197, 1255–1267.
- Yuan, Y.H.; Hove, T.; Ten; Olle The, F.; Slors, J.F.M.; Van Deventer, S.J.H.; Te Velde, A.A. Chemokine Receptor CXCR3 Expression in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2001, 7, 281–286.
- Agace, W.W.; Roberts, A.I.; Wu, L.; Greineder, C.; Ebert, E.C.; Parker, C.M. Human Intestinal Lamina Propria and Intraepithelial Lymphocytes Express Receptors Specific for Chemokines Induced by Inflammation. Eur. J. Immunol. 2000, 30, 819–826.
- Rojo, Ó.P.; San Román, A.L.; Arbizu, E.A.; Martínez, A.D.L.H.; Sevillano, E.R.; Martínez, A.A. Serum Lipopolysaccharide-Binding Protein in Endotoxemic Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2007, 13, 269–277.
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A Single-Cell Survey of the Small Intestinal Epithelium. Nature 2017, 551, 333–339.
- Lloyd, C.M.; Saglani, S. Epithelial Cytokines and Pulmonary Allergic Inflammation. Curr. Opin. Immunol. 2015, 34, 52–58.
- Kouzaki, H.; Tojima, I.; Kita, H.; Shimizu, T. Transcription of Interleukin-25 and Extracellular Release of the Protein Is Regulated by Allergen Proteases in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 741–750.
- Angkasekwinai, P.; Park, H.; Wang, Y.H.; Wang, Y.H.; Seon, H.C.; Corry, D.B.; Liu, Y.J.; Zhu, Z.; Dong, C. Interleukin 25 Promotes the Initiation of Proallergic Type 2 Responses. J. Exp. Med. 2007, 204, 1509–1517.
- Reynolds, J.M.; Lee, Y.H.; Shi, Y.; Wang, X.; Angkasekwinai, P.; Nallaparaju, K.C.; Flaherty, S.; Chang, S.H.; Watarai, H.; Dong, C. Interleukin-17B Antagonizes Interleukin-25-Mediated Mucosal Inflammation. Immunity 2015, 42, 692–703.
- Caruso, R.; Sarra, M.; Stolfi, C.; Rizzo, A.; Fina, D.; Fantini, M.C.; Pallone, F.; MacDonald, T.T.; Monteleone, G. Interleukin-25 Inhibits Interleukin-12 Production and Th1 Cell-Driven Inflammation in the Gut. Gastroenterology 2009, 136, 2270–2279.
- Borowczyk, J.; Shutova, M.; Brembilla, N.C.; Boehncke, W.H. IL-25 (IL-17E) in Epithelial Immunology and Pathophysiology. J. Allergy Clin. Immunol. 2021, 148, 40–52.
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 Induces IL-4, IL-5, and IL-13 and Th2-Associated Pathologies in Vivo. Immunity 2001, 15, 985–995.
- Tamachi, T.; Maezawa, Y.; Ikeda, K.; Kagami, S.I.; Hatano, M.; Seto, Y.; Suto, A.; Suzuki, K.; Watanabe, N.; Saito, Y.; et al. IL-25 Enhances Allergic Airway Inflammation by Amplifying a TH2 Cell-Dependent Pathway in Mice. J. Allergy Clin. Immunol. 2006, 118, 606–614.
- Koelink, P.J.; Overbeek, S.A.; Braber, S.; Morgan, M.E.; Henricks, P.A.J.; Roda, M.A.; Verspaget, H.W.; Wolfkamp, S.C.; Te Velde, A.A.; Jones, C.W.; et al. Collagen Degradation and Neutrophilic Infiltration: A Vicious Circle in Inflammatory Bowel Disease. Gut 2014, 63, 578–587.
- O’Reilly, P.; Jackson, P.L.; Noerager, B.; Parker, S.; Dransfield, M.; Gaggar, A.; Blalock, J.E. N-Alpha-PGP and PGP, Potential Biomarkers and Therapeutic Targets for COPD. Respir. Res. 2009, 10, 38.
- Keely, S.; Campbell, E.L.; Baird, A.W.; Hansbro, P.M.; Shalwitz, R.A.; Kotsakis, A.; McNamee, E.N.; Eltzschig, H.K.; Kominsky, D.J.; Colgan, S.P. Contribution of Epithelial Innate Immunity to Systemic Protection Afforded by Prolyl Hydroxylase Inhibition in Murine Colitis. Mucosal Immunol. 2013, 7, 114–123.
- Mateer, S.W.; Mathe, A.; Bruce, J.; Liu, G.; Maltby, S.; Fricker, M.; Goggins, B.J.; Tay, H.L.; Marks, E.; Burns, G.; et al. IL-6 Drives Neutrophil-Mediated Pulmonary Inflammation Associated with Bacteremia in Murine Models of Colitis. Am. J. Pathol. 2018, 188, 1625–1639.
- Liu, G.; Mateer, S.W.; Hsu, A.; Goggins, B.J.; Tay, H.; Mathe, A.; Fan, K.; Neal, R.; Bruce, J.; Burns, G.; et al. Platelet Activating Factor Receptor Regulates Colitis-Induced Pulmonary Inflammation through the NLRP3 Inflammasome. Mucosal Immunol. 2019, 12, 862–873.
- Mitchell, M.J.; Lin, K.S.; King, M.R. Fluid Shear Stress Increases Neutrophil Activation via Platelet-Activating Factor. Biophys. J. 2014, 106, 2243–2253.
- Liu, Y.; Wang, X.Y.; Yang, X.; Jing, S.; Zhu, L.; Gao, S.H. Lung and Intestine: A Specific Link in an Ulcerative Colitis Rat Model. Gastroenterol. Res. Pract. 2013, 2013, 124530.
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664.
- López-Serrano, P.; Pérez-Calle, J.L.; Pérez-Fernández, M.T.; Fernández-Font, J.M.; Boixeda De Miguel, D.; Fernández-Rodríguez, C.M. Environmental Risk Factors in Inflammatory Bowel Diseases. Investigating the Hygiene Hypothesis: A Spanish Case–Control Study. Scand. J. Gastroenterol. 2010, 45, 1464–1471.
- Birzele, L.T.; Depner, M.; Ege, M.J.; Engel, M.; Kublik, S.; Bernau, C.; Loss, G.J.; Genuneit, J.; Horak, E.; Schloter, M.; et al. Environmental and Mucosal Microbiota and Their Role in Childhood Asthma. Allergy 2016, 72, 109–119.
- Stefka, A.T.; Feehley, T.; Tripathi, P.; Qiu, J.; McCoy, K.; Mazmanian, S.K.; Tjota, M.Y.; Seo, G.Y.; Cao, S.; Theriault, B.R.; et al. Commensal Bacteria Protect against Food Allergen Sensitization. Proc. Natl. Acad. Sci. USA 2014, 111, 13145–13150.
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial Exposure during Early Life Has Persistent Effects on Natural Killer T Cell Function. Science 2012, 336, 489–493.
- Hugot, J.P. CARD15/NOD2 Mutations in Crohn’s Disease. Ann. N. Y. Acad. Sci. 2006, 1072, 9–18.
- Biank, V.; Broeckel, U.; Kugathasan, S. Pediatric Inflammatory Bowel Disease: Clinical and Molecular Genetics. Inflamm. Bowel Dis. 2007, 13, 1430–1438.
- Salem, M.; Seidelin, J.B.; Rogler, G.; Nielsen, O.H. Muramyl Dipeptide Responsive Pathways in Crohn’s Disease: From NOD2 and Beyond. Cell. Mol. Life Sci. 2013, 70, 3391–3404.
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s Disease: NOD2, Autophagy and ER Stress Converge. Gut 2011, 60, 1580–1588.
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schäffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) Mutations in Crohn’s Disease Are Associated with Diminished Mucosal Alpha-Defensin Expression. Gut 2004, 53, 1658–1664.
- Kinose, D.; Ogawa, E.; Hirota, T.; Ito, I.; Kudo, M.; Haruna, A.; Marumo, S.; Hoshino, Y.; Muro, S.; Hirai, T.; et al. A NOD2 Gene Polymorphism Is Associated with the Prevalence and Severity of Chronic Obstructive Pulmonary Disease in a Japanese Population. Respirology 2012, 17, 164–171.
- Scharl, M.; Rogler, G. Inflammatory Bowel Disease: Dysfunction of Autophagy? Dig. Dis. 2012, 30, 12–19.
- Orchard, T.R.; Chua, C.N.; Ahmad, T.; Cheng, H.; Welsh, K.I.; Jewell, D.P. Uveitis and Erythema Nodosum in Inflammatory Bowel Disease: Clinical Features and the Role of HLA Genes. Gastroenterology 2002, 123, 714–718.
- Roussomoustakaki, M.; Satsangi, J.; Welsh, K.; Louis, E.; Fanning, G.; Targan, S.; Landers, C.; Jewell, D.P. Genetic Markers May Predict Disease Behavior in Patients with Ulcerative Colitis. Gastroenterology 1997, 112, 1845–1853.
More
Information
Subjects:
Pediatrics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
20 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No