Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Harrison Rudd | -- | 2182 | 2022-07-06 01:02:59 | | | |
| 2 | Catherine Yang | Meta information modification | 2182 | 2022-07-13 03:27:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rudd, H.; Toborek, M. HIV Infection within the CNS. Encyclopedia. Available online: https://encyclopedia.pub/entry/25068 (accessed on 24 July 2026).
Rudd H, Toborek M. HIV Infection within the CNS. Encyclopedia. Available at: https://encyclopedia.pub/entry/25068. Accessed July 24, 2026.
Rudd, Harrison, Michal Toborek. "HIV Infection within the CNS" Encyclopedia, https://encyclopedia.pub/entry/25068 (accessed July 24, 2026).
Rudd, H., & Toborek, M. (2022, July 12). HIV Infection within the CNS. In Encyclopedia. https://encyclopedia.pub/entry/25068
Rudd, Harrison and Michal Toborek. "HIV Infection within the CNS." Encyclopedia. Web. 12 July, 2022.
Copy Citation
HIV can traverse the BBB using a Trojan horse-like mechanism. Hidden within infected immune cells, HIV can infiltrate the highly safeguarded CNS and propagate disease. Once integrated within the host genome, HIV becomes a stable provirus, which can remain dormant, evade detection by the immune system or antiretroviral therapy (ART), and result in rebound viraemia. As ART targets actively replicating HIV, has low BBB penetrance, and exposes patients to long-term toxicity, further investigation into novel therapeutic approaches is required.
HIV
antiretroviral therapy
blood-brain barrier
1. The Blood-Brain Barrier
The blood-brain barrier (BBB) is the crucial anatomic and biochemical interface responsible for regulating the microenvironments between peripheral circulation and the central nervous system (CNS) [1][2][3]. These highly regulated microenvironments are required for neural signaling and the maintenance of homeostasis within the CNS. Additional barriers including the blood–cerebrospinal fluid (CSF) barrier and the arachnoid barrier provide additional supportive functions in maintaining CNS homeostasis, but are not as crucial nor do they occupy as large of a surface area as the BBB. As such, the BBB is at the front line of defense in protecting the highly safeguarded CNS from the entrance of toxins and pathogens, including HIV and medications such as ART, adding an element of challenge to drug discovery and design.
At the BBB, a monolayer of cerebral microvascular endothelial cells (CMECs) forms the framework of capillary walls, which are interlocked by tight junctions (TJs) made up of proteins including claudin-5, occludin, and submembranous zona occludnes-1 (ZO-1). These TJs facilitate the regulation of BBB and CNS homeostasis by linking together CMECs, preventing the passage of many paracellular molecules into the brain parenchyma, while also providing a cytoskeletal matrix of intercellular protein filaments arranged as a series of membranous and submembranous barricades that enable the structural and functional maintenance of barrier integrity. CMECs are peripherally surrounded by a basement membrane (basal lamina), pericytes, astrocytic end-feet, and neurons, which together comprise the neurovascular unit and serve to strengthen barrier function and integrity at the BBB [3]. Pericytes and astrocytes have important roles in maintaining structural integrity at the BBB as they can modulate levels of TJ protein expression and vesicle trafficking in CMECs [4] and contribute to various aspects of CMEC phenotype, including development, proliferation, migration, and survival [2][4]. Interestingly, pericytes are also able to regulate the expression of BBB-specific genes in CMECs, influencing overall BBB integrity [5].
There are several pathways that restrict the entry of drugs into the CNS. Molecules that can diffuse or be transported through the endothelium, including ART, can be actively removed via efflux pumps including P-glycoprotein, multidrug resistance proteins, and organic anion transporters [6]. This provides a challenge for ART in reaching therapeutic concentrations within the CNS, allowing for the possibility of rebound viraemia. It is, however, essential to note that several factors can modulate the expression of these transport proteins, such as inflammatory, genetic, and drug-induced interactions [6], which can result in increased transport of ART across the brain endothelium, leading to increased toxic exposure. Of particular interest is the role of P-glycoprotein in limiting entry into the CNS for ARVds. Substantial effort has been made to dissect the regulatory mechanisms modulating the expression and/or activity of this protein. Exposure of capillaries to low levels of proinflammatory factors, such as lipopolysaccharide (LPS), tumor necrosis factor (TNF)-α, or endothelin-1 (ET-1), was demonstrated to cause a rapid loss of P-glycoprotein transport function with no change in protein expression. On the other hand, a prolonged exposure to proinflammatory factors, including TNF-α, had an opposing effect, i.e., upregulating P-glycoprotein expression via complex mechanisms that shared common signaling elements, such as TNF receptor 1, endothelin receptors, protein kinase C, and nuclear factor-κB (NF-κB) [7]. The role of inflammatory factors in the modulation of P-glycoprotein activity has been confirmed in several literature reports (reviewed in [8]). Interestingly, P-glycoprotein is also involved in the immune inflammatory response in the CNS by regulating microglia activation and mediating immune cell migration [9].
Hydrophobicity and low molecular weight are positively correlated with BBB penetration [6][10]; however, efflux pumps may still actively remove these substances from the brain parenchyma. The inability of certain antiretroviral drugs (ARVds) to reach therapeutic concentrations within the CNS may play a role in the potential for rebound viraemia. Thus, future methods of drug delivery must be investigated and optimized to bypass the classical diffusion and transport mechanisms of ART across the BBB.
2. A Trojan Horse Mechanism for HIV Infection of the CNS
HIV attacks the immune system by infecting and eliminating cells that express the CD4 receptor (CD4+ cells) and coreceptors, including the C-C motif receptor 5 (CCR5) and C-X-C motif receptor 4 (CXCR4). The HIV genome can integrate into the host genome of many cell types; however, there are two major cellular reservoirs: CD4+ T lymphocytes and macrophages. CD4+ T cells are crucial for combating infection and maintaining immune responses, homeostasis, and memory, and as such, are associated with several inflammatory and autoimmune diseases [11]. Macrophages are derived from monocytes and myeloid cells of hematopoietic origin [9]. In the CNS, microglia and partially pericytes are cells of myeloid origin. These cells, along with astrocytes, can all be directly infected by HIV [12][13][14][15][16]. Indeed, a 12 h incubation period with two strains of HIV resulted in the cellular entry of HIV and low-level replication of HIV in human brain pericytes, astrocytes, and CMECs [17].
HIV can infiltrate the CNS early in the course of infection. HIV evades detection by the immune system primarily by using HIV+ CD4+ T cells and cells of the monocytic lineage in a Trojan horse approach to traverse the BBB [17] (Figure 1). The free virus is also able to cross the BBB through TJ openings that can result from HIV-induced dysfunction of CMECs [1]. In addition, HIV+ pericytes were shown to stimulate dysregulation of BBB integrity via decreased TJ protein expression [16]. This HIV-induced increase in BBB permeability can lead to the activation of microglial cells and uncontrolled migration of immune cells into the CNS, which are capable of causing neuroinflammation, loss of neural tissue, and infection due to the influx of pathogens [18]. In addition, studies have shown that CMECs can undergo apoptosis during HIV infection [1][16], which increases BBB permeability and can promote the infiltration of HIV+ cells and virions into the CNS. Intriguingly, HIV-specific proteins, such as transactivator of transcription (Tat), are capable of inducing CMEC dysfunction and subsequent BBB dysregulation [19][20][21], enhancing the infiltration of HIV across the BBB. The entry of HIV into the CNS can result in neuropathological dysfunctions ranging from sub-clinical and minor cognitive impairments or motor deficits to HIV-associated neurocognitive disorders (HAND), including dementia [22]. ART administration is negatively correlated to TJ protein expression and BBB permeability [4][18][23], illustrating the need for drug design to maximize the efficiency of BBB crossing and to overcome toxicities associated with the administration of ART.
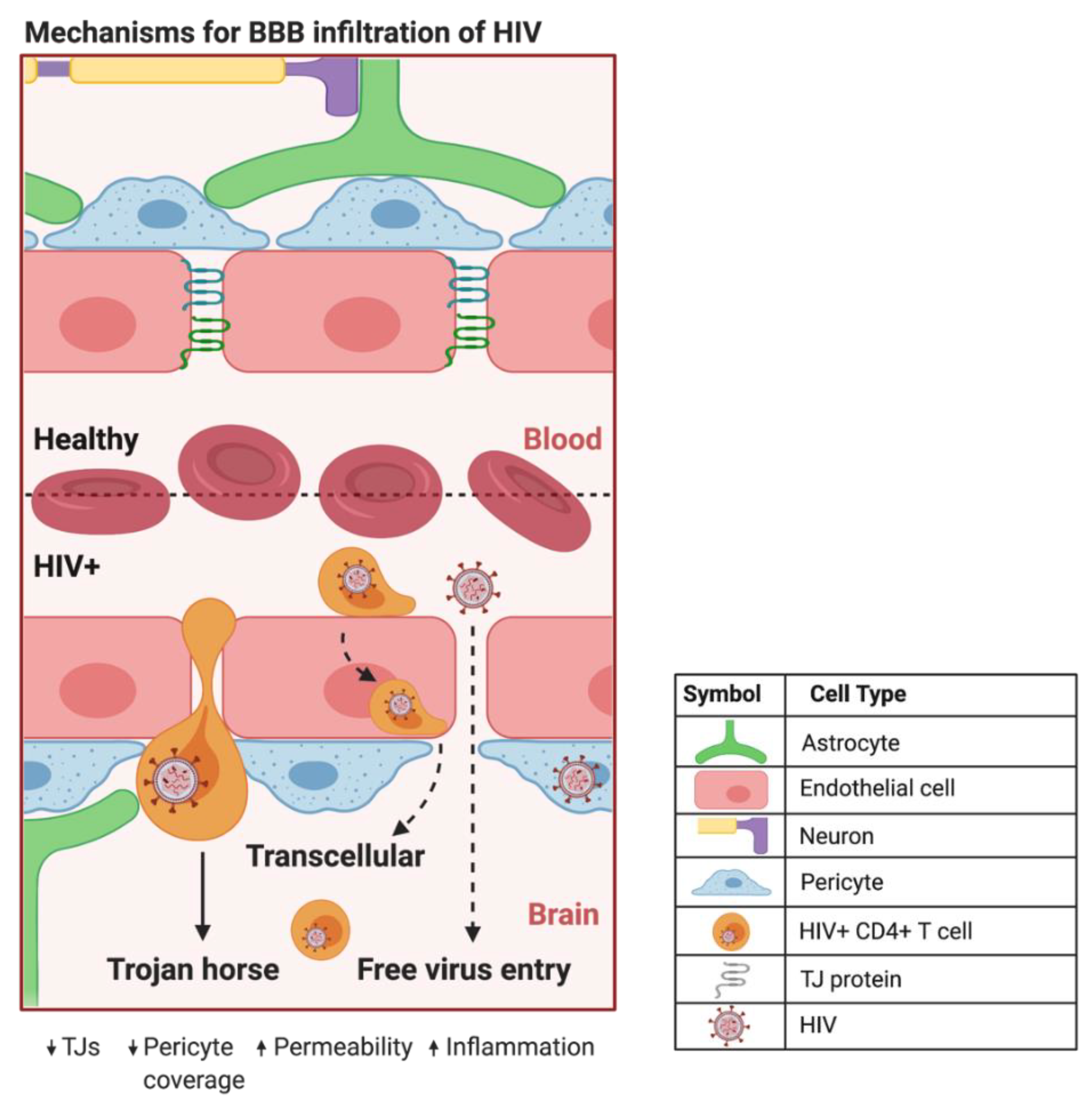
Figure 1. Proposed mechanisms of BBB infiltration of HIV. At the HIV+ BBB, infected CD4+ T cells and monocytes can cross by several proposed mechanisms. The predominate method centers around HIV using infected CD4+ T cells and monocytes as a Trojan horse to paracellularly infiltrate brain parenchyma. HIV+ monocytes can also transcellularly pass through CMECs at the BBB. As HIV infection progresses in the CNS, increased BBB permeability and decreased expression of TJ proteins can provide a pathway for HIV to paracellularly invade the brain parenchyma. Created with BioRender.com. Abbreviations: HIV+ = human immunodeficiency virus-infected; TJs = tight junctions.
3. Elusive Latent Proviral Reservoirs within the CNS
The integration of reverse-transcribed viral DNA into the host genome is a crucial step in propagating both the active and dormant forms of HIV (Figure 2). Once integrated, the proviral DNA serves as the transcriptionally competent viral unit and the central source of viral protein production. The gene expression of HIV is controlled by promoter and enhancer sequences where transcription factors, including NF-κB, can bind, promoting RNA polymerase II activity, ultimately resulting in increased virus-specific protein levels [24]. Transcriptional inactivity of the HIV proviral DNA results in the latent proviral stage of HIV, where the virus can remain dormant in the host genome as a transcriptionally incompetent reservoir for later reactivation.
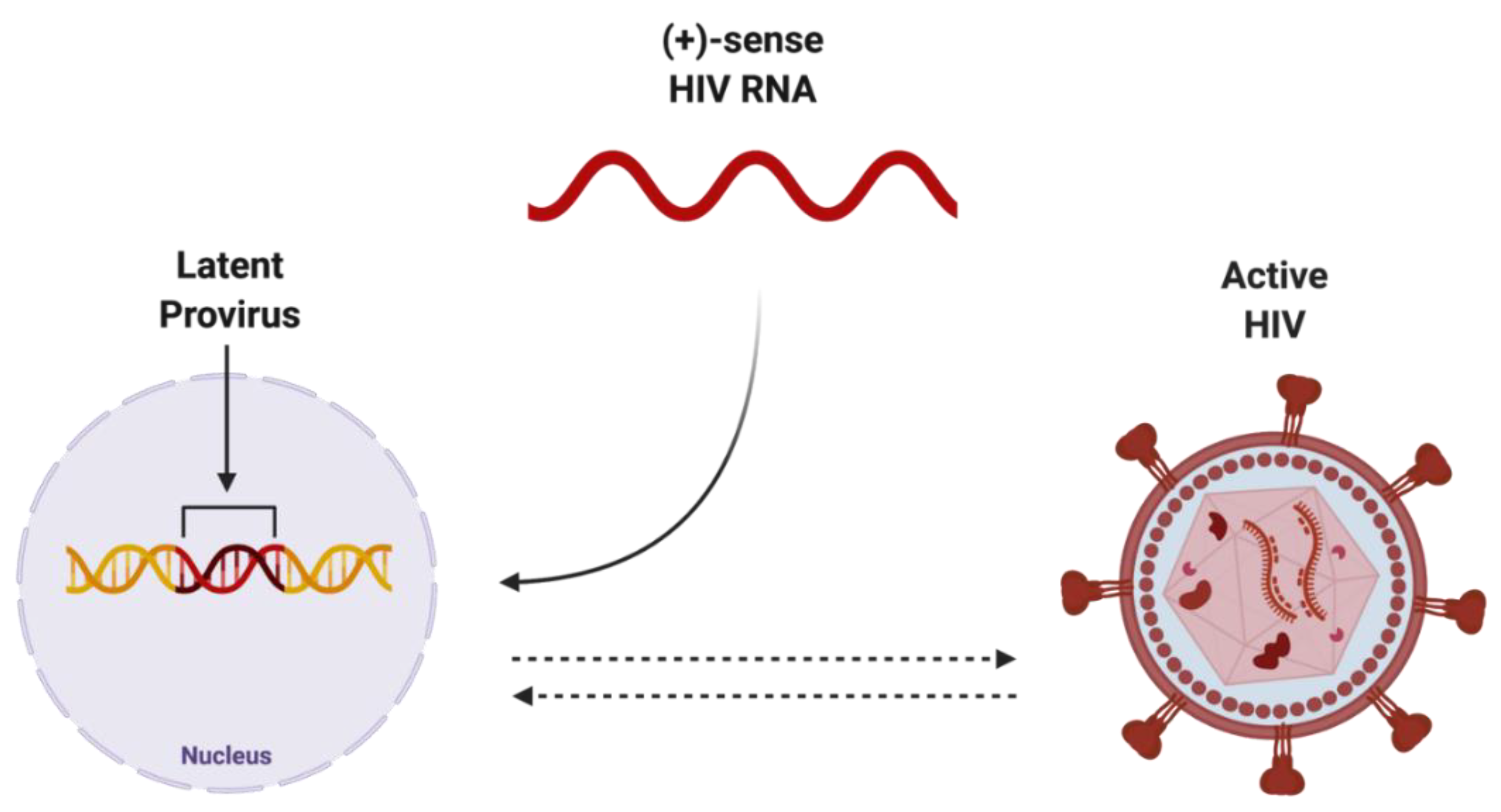
Figure 2. Potential endpoints of positive-sense HIV RNA after integration into host genome. Once integrated into the host genome, (+)-sense HIV RNA can persist as either latent provirus, which is capable of being reactivated, or actively replicating HIV, which can be deactivated. Created with BioRender.com. Abbreviations: (+)-sense = positive-sense; HIV = human immunodeficiency virus; RNA = ribonucleic acid.
The HIV provirus can exist in three forms: latent, which is transcriptionally silent; intact, producing active virions; or defective, producing viral proteins but not able to successfully replicate [25] (Figure 3). Intact and latent HIV proviral reservoirs have the potential to cause rebound viraemia, whereas defective provirus does not. It is important to note that while defective HIV provirus is not replication-competent, these malfunctioning viral DNA sequences can produce viral HIV proteins, which can propagate pathogenesis. Furthermore, cells latently infected with HIV can release exosomes containing viral mRNA and protein, hijacking intercellular communication networks as a means to reactivate latent reservoirs, transmit infection, and further disease development [26], presenting another target required to fully eradicate infection with HIV.
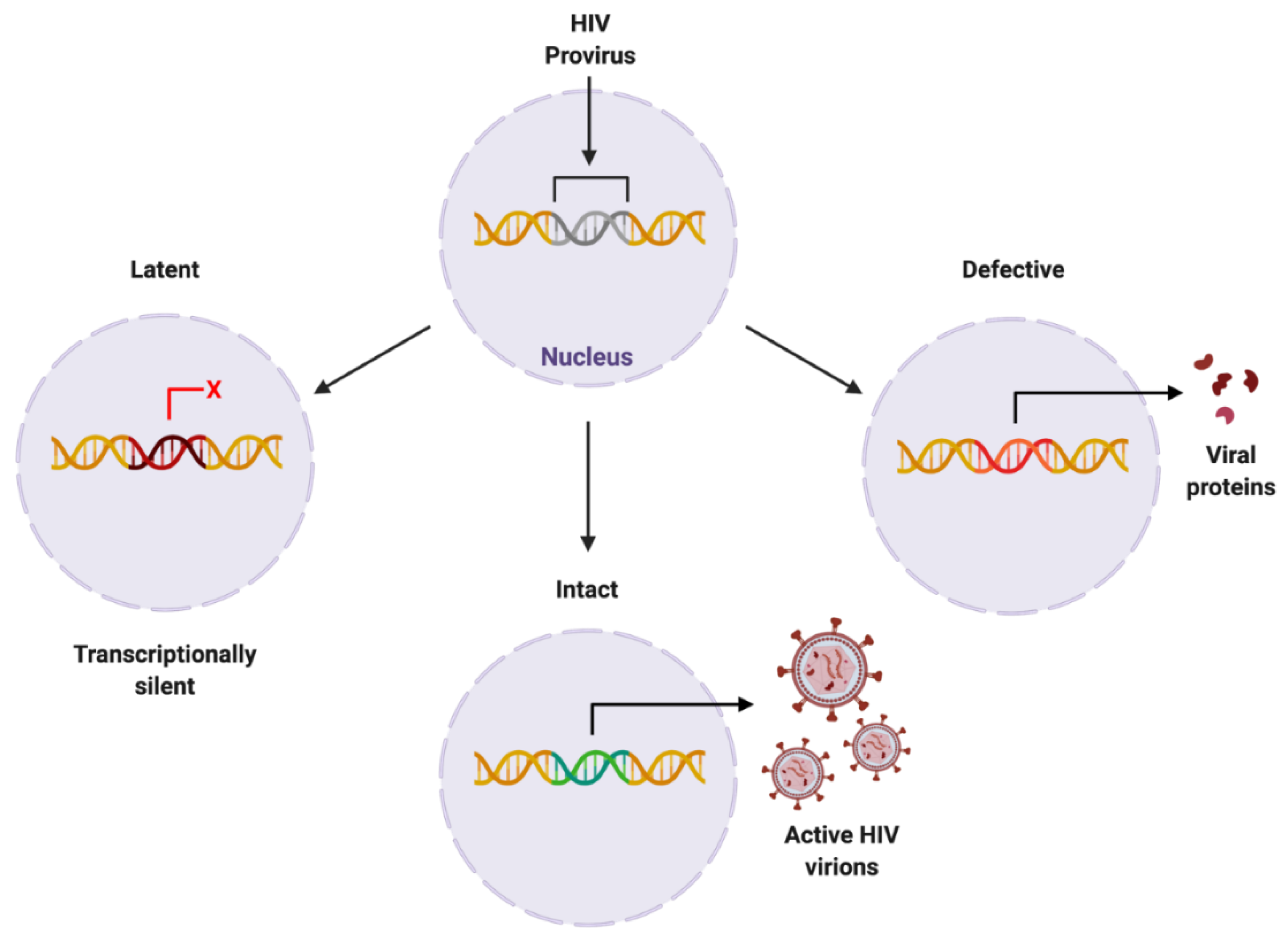
Figure 3. Proposed forms of HIV proviral reservoir. HIV provirus can persist in three forms: latent, being transcriptionally silent; intact, producing active HIV virions; or defective, containing genetic mutations resulting in viral protein synthesis. Created with BioRender.com. Abbreviations: HIV = human immunodeficiency virus.
HIV can exist as a latent proviral reservoir in several cell types, namely CD4+ T cells and cells of monocytic lineage. Cells that are latently infected with HIV provirus can evade detection by the immune system and may be replicated via the homeostatic proliferation of their host cell [25]. Although microglial cells are the primary reservoir cell type within the CNS [27], there is novel evidence depicting astrocytes and pericytes as constituents of these dormant HIV cellular reservoirs [13][14]. For example, integrated viral DNA has been discovered in microglia and pericytes within the CNS tissue of post-mortem HIV+ patients [12][25], illustrating the likelihood of myeloid-derived reservoir sites within the brain. Intriguingly, novel research indicates a key role for neurons, as opposed to non-neuronal cell lines, in the stimulation of HIV latency in microglia [28]. In addition, neurons can prevent the emergence of active HIV from latency and neuronal damage can induce replication and activation of HIV [28]. As cells of the myeloid lineage are long-lasting and recent investigation has illustrated both inductive and preventative roles for neurons in HIV latency, it is crucial to further investigate the functional properties of HIV latency.
Activating the transcriptionally silent, latent HIV proviral reservoir can be achieved with the use of histone deacetylase inhibitors. Histone deacetylase inhibitors promote the acetylation of histones and consequential chromatin relaxation, facilitating the accessibility of transcription factors to DNA and enabling transcription of the viral genome via RNA polymerase II recruitment. For example, pericytes in the latent stage of HIV infection that were treated with histone deacetylase inhibitors and tumor necrosis factor (TNF) exhibited a significant increase in HIV-1 RNA and HIV p24 protein production, illustrating that pericytes can alternate between the latent and active viral stages [12]. The mechanisms underlying HIV proviral transcriptional silencing and reactivation are not yet fully understood. Recent investigation has revealed a method of measuring and discerning the intact versus defective proviral HIV genome [29], which is a crucial step toward curing HIV infection. Specific targeting of the latent proviral reservoir remains the central obstacle in achieving complete viral eradication from HIV+ individuals.
Perivascular spaces in the CNS also contain populations of cells capable of harboring HIV. In a macaque model, perivascular macrophages and microglia were shown to harbor SIV genomes, which could be reactivated, even after observed antiretroviral therapy suppression [30]. While there is still debate regarding the role that macrophages play in active viral reservoirs of HIV, findings in mice confirm the possible importance of this cell type. Indeed, studies indicated that HIV persists in humanized myeloid-only mice independent of other possible reservoir-capable cell types, such as T-cells, supporting the role of macrophages in HIV replication and formation of viral reservoirs. This mouse model is generated by transplanting CD34+ hematopoietic stem cells into immunodeficient nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, which are characterized by an absence of functional T and B cells [31][32]. Furthermore, there is mounting evidence that macrophages play an important role in their susceptibility to HIV even after ART initiation (reviewed in [10]).
Evidence indicates that viral entry can occur through the choroid plexus [33][34]. It is well-known that resident macrophages, i.e., the cells that frequently become infected with HIV in the CNS, can line the epithelium of the choroid plexus [35]. As a separate dynamic reservoir for HIV accumulation, the choroid plexus provides a possible path for neuro-invasion events and a conduit for future ART drug delivery. It should also be noted that HIV trafficking via the choroid plexus barrier is coordinated by the high amount of multidrug resistance proteins and P-glycoprotein expressed on the surface [36]. Paradoxically, the P-glycoprotein pump is oriented in a way that opposes the action of P-glycoprotein efflux transporter located in the BBB, whereby it prevents substrates and other molecules from escaping the CSF. This complex relationship further accentuates CNS and BBB homeostasis in trafficking therapeutics to the CNS (reviewed in [10]).
References
- Toborek, M.; Lee, Y.W.; Flora, G.; Pu, H.; András, I.E.; Wylegala, E.; Hennig, B.; Nath, A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell. Mol. Neurobiol. 2005, 25, 181–199.
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236.
- Suzuki, Y.; Nagai, N.; Umemura, K. A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front. Cell. Neurosci. 2016, 10, 2.
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23.
- Kurmann, L.; Okoniewski, M.; Dubey, R.K. Transcryptomic analysis of human brain -microvascular endothelial cell driven changes in -vascular pericytes. Cells 2021, 10, 1784.
- Bertrand, L.; Velichkovska, M.; Toborek, M. Cerebral vascular toxicity of antiretroviral therapy. J. Neuroimmune Pharmacol. 2019, 16, 74–89.
- Miller, D.S.; Bauer, B.; Hartz, A.M. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209.
- Deng, X.; Xie, Y.; Chen, Y. Effect of Neuroinflammation on ABC Transporters: Possible Contribution to Refractory Epilepsy. CNS Neurol. Disord. Drug Targets 2018, 17, 728–735.
- Huang, L.; Li, B.; Li, X.; Liu, G.; Liu, R.; Guo, J.; Xu, B.; Li, Y.; Fang, W. Significance and Mechanisms of P-glycoprotein in Central Nervous System Diseases. Curr. Cancer Drug Targets 2019, 20, 1141–1155.
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The paradox of HIV blood-brain barrier penetrance and antiretroviral drug delivery deficiencies. Trends Neurosci. 2020, 43, 695–708.
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T cell development, localization, and function throughout life. Immunity 2018, 48, 202–213.
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 2019, 142, 502–511.
- Li, G.-H.; Henderson, L.; Nath, A. Astrocytes as an HIV reservoir: Mechanism of HIV infection. Curr. HIV Res. 2016, 14, 373–381.
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. HIV-1 target cells in the CNS. J. NeuroVirology 2014, 21, 276–289.
- András, I.E.; Toborek, M. HIV-1-induced alterations of Claudin-5 expression at the blood-brain barrier level. Methods Mol. Biol. 2011, 762, 355–370.
- Nakagawa, S.; Castro, V.; Toborek, M. Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J. Cell. Mol. Med. 2012, 16, 2950–2957.
- Kawamoto, H.; Minato, N. Myeloid cells. Int. J. Biochem. Cell Biol. 2004, 36, 1374–1379.
- Bertrand, L.; Dygert, L.; Toborek, M. Antiretroviral treatment with Efavirenz disrupts the blood-brain barrier integrity and increases stroke severity. Sci. Rep. 2016, 6, 39738.
- Zhong, Y.; Zhang, B.; Eum, S.Y.; Toborek, M. HIV-1 TAT triggers nuclear localization of ZO-1 via rho signaling and camp response element-binding protein activation. J. Neurosci. 2012, 32, 143–150.
- András, I.E.; Pu, H.; Tian, J.; Deli, M.A.; Nath, A.; Hennig, B.; Toborek, M. Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J. Cereb. Blood Flow Metab. 2005, 25, 1159–1170.
- Lee, Y.W.; Eum, S.Y.; Nath, A.; Toborek, M. Estrogen-mediated protection against HIV Tat protein-induced inflammatory pathways in human vascular endothelial cells. Cardiovasc. Res. 2004, 63, 139–148.
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986.
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and art. FEBS J. 2018, 286, 1256–1270.
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3.
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward curing HIV-1 infection in tissue reservoirs. J. Virol. 2020, 94, e00375-19.
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87.
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial cells: The main HIV-1 reservoir in the brain. Front. Cell. Infect. Microbiol. 2019, 9, 362.
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249.
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125.
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165.
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643.
- Gu, C.J.; Borjabad, A.; Hadas, E.; Kelschenbach, J.; Kim, B.H.; Chao, W.; Arancio, O.; Suh, J.; Polsky, B.; McMillan, J.; et al. EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLoS Pathog. 2018, 14, e1007061.
- Delery, E.C.; MacLean, A.G. Culture Model for Non-human Primate Choroid Plexus. Front. Cell. Neurosci. 2019, 13, 396.
- Meeker, R.B.; Williams, K.; Killebrew, D.A.; Hudson, L.C. Cell trafficking through the choroid plexus. Cell Adhes. Migr. 2012, 6, 390–396.
- Burkala, E.J.; He, J.; West, J.T.; Wood, C.; Petito, C.K. Compartmentalization of HIV-1 in the central nervous system: Role of the choroid plexus. AIDS 2005, 19, 675–684.
- Rao, V.V.; Dahlheimer, J.L.; Bardgett, M.E.; Snyder, A.Z.; Finch, R.A.; Sartorelli, A.C.; Piwnica-Worms, D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 3900–3905.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
13 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No