Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Izabela Szymczak-Pajor | -- | 1844 | 2022-07-07 07:44:27 | | | |
| 2 | Vivi Li | Meta information modification | 1844 | 2022-07-08 03:47:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Szymczak-Pajor, I.; Wenclewska, S.; Śliwińska, A. Metabolic Action of Metformin. Encyclopedia. Available online: https://encyclopedia.pub/entry/24888 (accessed on 25 July 2026).
Szymczak-Pajor I, Wenclewska S, Śliwińska A. Metabolic Action of Metformin. Encyclopedia. Available at: https://encyclopedia.pub/entry/24888. Accessed July 25, 2026.
Szymczak-Pajor, Izabela, Sylwia Wenclewska, Agnieszka Śliwińska. "Metabolic Action of Metformin" Encyclopedia, https://encyclopedia.pub/entry/24888 (accessed July 25, 2026).
Szymczak-Pajor, I., Wenclewska, S., & Śliwińska, A. (2022, July 07). Metabolic Action of Metformin. In Encyclopedia. https://encyclopedia.pub/entry/24888
Szymczak-Pajor, Izabela, et al. "Metabolic Action of Metformin." Encyclopedia. Web. 07 July, 2022.
Copy Citation
Metformin, a cheap and safe biguanide derivative, due to its ability to influence metabolism, is widely used as a first-line drug for type 2 diabetes (T2DM) treatment.
metformin
hepatic gluconeogenesis
lipid metabolism
1. Introduction
Currently, the incidence of type 2 diabetes (T2DM) is becoming an epidemic, and the treatment of complications caused by chronic hyperglycemia is extremely economically burdensome. Chronic hyperglycemia exerts a direct toxic effect on different cell types, including pancreatic β-cells and vascular endothelial cells. Specifically, in the insulin resistance state preceding symptomatic T2DM, prolonged hyperglycemia contributes to oxidative stress that is highly dangerous to β-cells. Moreover, as a result of increased secretion of insulin, β-cells become exhausted and die. Thus, secretion of insulin is disturbed. In turn, vascular endothelial cells are particularly sensitive to hyperglycemia since they transport glucose in an insulin-independent manner, and intracellular glucose concentration is proportional to its blood concentration. Thus, endothelial cells are directly exposed to the toxic effect of high glucose and related oxidative stress, which leads to micro- and macro-vasculature dysfunction, initiating the development of diabetes complications in multiple organs that significantly affect the length and quality of life. It was demonstrated that metformin action is limited not only to the decrease in hyperglycemia, but also to a delay in the development of diabetic complications [1][2][3]. The mechanisms of metformin’s action are complex and associated with multiple targets in the body [4][5]. Therefore, the aim of the entry was to provide updated knowledge concerning the molecular and biochemical actions of metformin involved in metabolism regulation.
2. The Fate of Metformin in the Human Body
After oral ingestion, metformin is absorbed by proximal small-intestine enterocytes. It has been proposed that passive diffusion is responsible for 50% of metformin uptake in the intestine. This type of transport is conducted by paracellular and transcellular pathways. However, there is no agreement whether paracellular, transcellular, or both pathways are engaged in metformin transport [6]. In addition to passive diffusion, the rest of the drug is transported by facilitated diffusion employing numerous transporters. The primary transporter involved in metformin’s absorption is plasma membrane monoamine transporter (PMAT) present on polarized enterocyte apical membranes. The affinity of PMAT to metformin determined by Michaelis constant (Km) is equal to 1.32 mM [7]. Another transporter found on the apical membrane of enterocytes participating in metformin absorption is organic cation transporter 3 (OCT3) that possess a Km of 1.10 mM. Due to the fact that OCT3 possesses a lower Km for metformin, its affinity to the drug is higher as compared to PMAT. This was confirmed by Chen et al., who reported that the deletion of OCT3 evoked decreased metformin bioavailability and circulating level [8]. The transport of metformin into the portal vein also occurs through OCT1 found on the basolateral membrane of enterocytes [9]. Several other transporters such as serotonin transporter (SERT), thiamine transporter 2 (THTR2), and carnitine/organic cation transporter 1 (OCTN1) participating in the intestinal absorption of metformin have been also identified. However, their precise role in metformin absorption is not fully known.
It was observed that the drug is undetectable in the plasma for 24 h after oral administration of single dose, and its half-life of elimination is equal to 7.2 h [10]. Plasma metformin levels in the portal vein range from 40 to 70 µM in animals after administration of a therapeutic dose. As a result of the transport of metformin with the blood, the drug is delivered directly to the liver, where its uptake is mediated by OCT1 and OCT3, achieving 3–5 times higher concentrations than in portal vein [11]. OCT1 and OCT3 are key transporters that take up the drug, since their knockout evokes significantly lower hepatic metformin accumulation and reduced suppression of glucose production [8][12][13]. As a result of hepatic uptake of metformin, its plasma concentration drops to 10–40 µM in both humans and animals [14]. The excretion of metformin from hepatocytes to circulation occurs through multidrug and toxin extrusion 1 (MATE1), and MATE inhibition causes the accumulation of the drug in the liver [15].
Metformin is not metabolized by the liver; however, MATE1 expressed in hepatocytes is involved in elimination of unchanged drug with the bile or in transport of metformin with the blood to kidney [10]. Renal excretion of the drug in unchanged form by tubular secretion into urine is the major pathway of metformin clearance [16][17]. The transport of metformin to the kidney is mediated by MATE1, MATE2, and OCT2. The latter is a key transporter for uptake of the drug by renal epithelial cells (Km of 0.99 mM) [18]. In turn, MATE2 (Km of 1.05 mM) and MATE1 (Km of 0.23 mM) participate in metformin secretion from the tubule cells into the urine. Furthermore, MATE1 is also involved in the secretion of metformin into the bile [17][19][20][21]. The fate of metformin in the human body is presented in Figure 1.
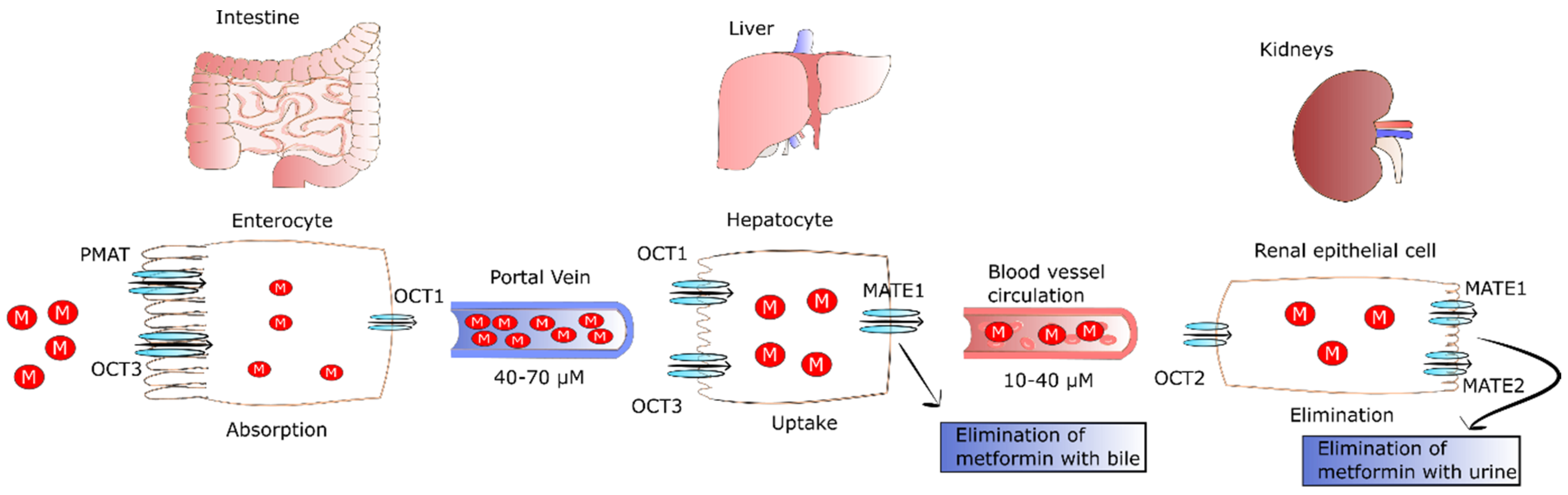
Figure 1. The fate of metformin in the human body. After oral ingestion, 50% of metformin is absorbed by passive diffusion, and the rest of the drug is transported by facilitated diffusion via PMAT and OCT1 transporters in intestinal enterocytes. Then, the drug leaves enterocytes via OCT1 and is transported to the liver via the portal vein where it reaches concentrations of 40–70 µM. Metformin enters the liver via OCT1 and OCT3 where it inhibits gluconeogenesis. The drug is not metabolized by the liver, but MATE1 expressed in hepatocytes participates in elimination of unchanged drug with the bile or in its transport with the blood to kidney. Then, metformin enters renal epithelial cells via OCT2. Next, the drug is secreted by renal MATE1 and MATE2 in unchanged form and eliminated with urine. Abbreviations: M, metformin; PMAT, plasma membrane monoamine transporter; OCT1, organic cation transporter 1; OCT2, organic cation transporter 2; OCT3, organic cation transporter 3; MATE 1, multidrug and toxin extrusion protein 1; MATE2, multidrug and toxin extrusion protein 2.
Comparing the Km of MATE1 and MATE2 of renal cells, one can observe that MATE1 has higher affinity to metformin than MATE2. Gan et al. revealed that MATE1 deletion evoked systemic elevation of metformin and lactic acidosis, the major side-effect of the drug. The pathophysiology of metformin-induced lactic acidosis is likely because of suppression of gluconeogenesis via blocking of pyruvate carboxylase (PC) [22]. The lack of PC activity results in the plasma accumulation of pyruvate that is next changed into lactic acid via lactate dehydrogenase (LDH), triggering its increased plasma concentration. LDH is an enzyme catalyzing the conversion of pyruvate into lactate and the reverse reaction that require NAD+ reduction and NADH oxidation, respectively. Hepatic transformation of lactate into pyruvate and muscle-specific conversion of pyruvate into lactate are closely related processes known as the Cori cycle or lactic acidic cycle that comprises gluconeogenesis and glycolysis. In the Cori cycle, anaerobic glycolysis-derived lactate is transported from the muscle to the liver where it is transformed into glucose. In turn, glucose returns to the muscles where it is metabolized to lactic acid [23][24][25]. Thus, the Cori cycle may be involved in metformin-induced lactic acidosis because the drug inhibits PC activity, leading to plasma accumulation of pyruvate. In turn, pyruvate participates in the Cori cycle and is transformed into lactic acid. At therapeutic concentrations of metformin, lactate is changed back to glucose in the Cori cycle. Conversely, metformin accumulation as a result of improper elimination or excessive drug intake leads to reduced hepatic lactate uptake and lactic acidosis. However, lactic acidosis and hyperlactatemia do not develop in all patients with isolated overdose of metformin [26].
3. Metformin Regulates Lipid Metabolism
3.1. Metformin Decreases the Secretion of Lipids from Intestinal Epithelial Cells
T2DM patients present abnormal metabolism of lipids, leading to significantly increased coexistence of fatty liver and cardiovascular diseases. On the other hand, metformin improves lipid metabolism, thereby reducing the risk of fatty liver and cardiovascular complications. This effect is partially connected to the property of metformin to decrease the concentration of chylomicrons in T2DM patients [27]. Metformin, via activation of AMPK and GLP1, diminishes the synthesis of apoA-IV and apoB-48. These are crucial mediators of chylomicron synthesis and secretion, and they are elevated in T2DM patients. The decreased level of apoA-IV and apoB-48 and reduced synthesis of triglycerides caused by metformin lead to a lowering of chylomicron formation, as well as secretion by enterocytes [28][29][30][31]. In addition, metformin was found to diminish cholesterol level in the circulation via a reduction in the reabsorption of bile acids in the intestine and an increase in chylomicron clearance [32].
3.2. Metformin as an Enhancer of Oxidation of Fatty Acids in Adipose Tissue and Muscles
It was shown that metformin treatment of mice fed with HFD and T2DM patients exhibited loss of adipose tissue as a result of elevated uptake and utilization of fatty acids [33][34]. This led to a reduction in VLDL-TG level and lipid droplet content in BAT. Metformin pronouncedly elevates fatty-acid utilization and oxidative phosphorylation in the mitochondria via increasing the proteins involved in the mitochondrial respiratory chain. The drug was also reported to activate hormone-sensitive lipase (HSL) expression and phosphorylation of acetyl-coenzyme A carboxylase (ACC), AMPK, and HSL in differentiated adipocytes, thereby increasing lipolysis. It was also observed that metformin elevated both utilization and uptake of fatty acids in adipose tissue, which in turn may have been related to the decreasing in VLDL-TG and mass of adipose tissue in mice fed with HFD and patients [33][34]. Metformin-stimulated fibroblast growth factor 21 (FGF21) seems to be involved in the reduced mass of adipose tissue and elevation of fatty-acid oxidation in white adipocytes derived from obese mice. FGF21 is an important metabolic regulator participating in the control of lipolysis in WAT [35]. Wang et al. observed that metformin can suppress accumulation of fat via promoting fatty-acid oxidation in the skeletal muscle of ob/ob mice. Some genes engaged in fatty-acid oxidation and synthesis of acyl-CoA were downregulated including Ascl3, Ppard, Mlycd, and Acsbg1 [36]. The mechanisms by which metformin regulates metabolism of lipids in intestinal epithelial cells, muscles and adipose tissue are presented in Figure 2.
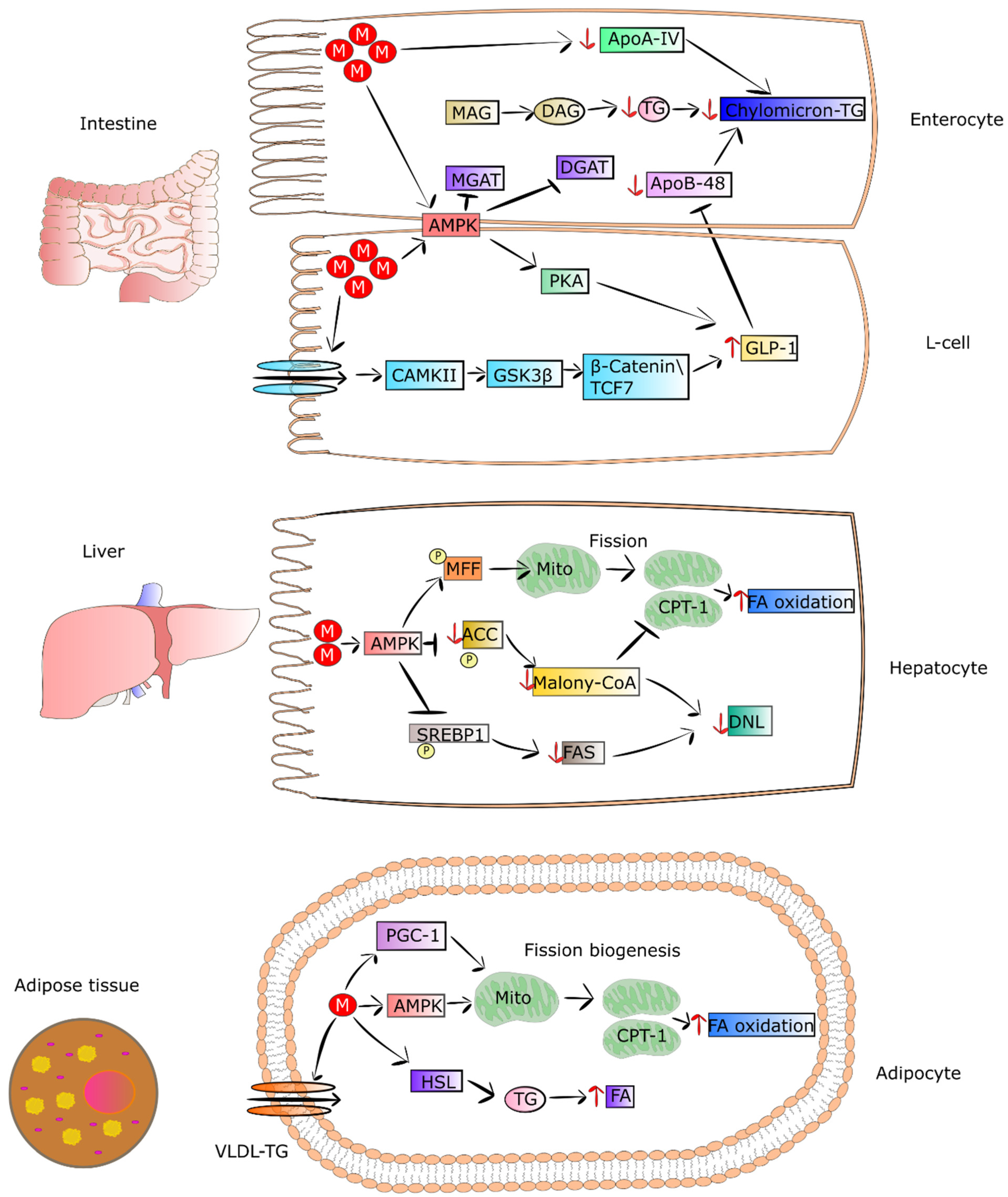
Figure 2. Metformin regulates the metabolism of lipids in enterocytes, L-cells, hepatocytes, and adipocytes. In the intestine, the drug initiates the expression of GLP-1 and its secretion via AMPK-independent and -dependent pathways in L cells. In enterocytes, metformin suppresses chylomicron storage and production via decreasing the levels of Apo-IV and Apo-48, as well as the synthesis of TG. In turn, in hepatocytes, metformin-dependent activated AMPK phosphorylates ACC and SREBP-1, leading to suppression of DNL and restoring CPT-1 activity. The drug also promotes the process of mitochondrial fission, leading to an increase in mitochondrial number and elevation of FA oxidation. The action of metformin in adipocytes contributes to increased uptake of FA and HSL activity, involving lipolysis. The drug also strengthens mitochondrial biogenesis via inducing PGC-1 signaling. Additionally, metformin activates AMPK, which also promotes mitochondrial fission. Lastly, the drug enhances oxidation of FA in adipocytes. Abbreviations: M, metformin; MAG, myelin-associated glycoprotein precursor; DAG, diacylglycerol; TG, triglycerides; ApoA-IV, apolipoprotein A-IV; ApoB-48, apolipoprotein B-48; MGAT, monoacylglycerol acyltransferase; DGAT, diglyceride acyltransferase; AMPK, 5’ adenosine monophosphate-activated protein kinase; PKA, protein kinase A; GLP-1, glucagon-like peptide-1; CAMPii, Ca2+/calmodulin-dependent protein kinase II; GSK-3β, glycogen synthase kinase-3 β; TCF7, transcription factor 7; MFF, mitochondrial fission factor; ACC, acetyl-CoA carboxylase; CPT1, carnitine palmitoyltransferase I; FA, fatty acid; FAS; DNL, de novo lipogenesis; SREBP1, sterol regulatory element-binding protein 1; malony-CoA, malonyl-coenzyme A; VLDL-TG, high-plasma very-low-density lipoprotein triglyceride; PGC1, peroxisome proliferation-activated receptor-gamma coactivator-1; HSL, hormone-sensitive lipase.
References
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S98–S110.
- Scheen, A.J.; Paquot, N. Metformin Revisited: A Critical Review of the Benefit-Risk Balance in at-Risk Patients with Type 2 Diabetes. Diabetes Metab. 2013, 39, 179–190.
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin Resistance: Review of the Underlying Molecular Mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161.
- King, P.; Peacock, I.; Donnelly, R. The UK Prospective Diabetes Study (UKPDS): Clinical and Therapeutic Implications for Type 2 Diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648.
- DeFronzo, R.A.; Goodman, A.M.; The Multicenter Metformin Study Group. Efficacy of Metformin in Patients with Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1995, 333, 541–549.
- Shirasaka, Y.; Seki, M.; Hatakeyama, M.; Kurokawa, Y.; Uchiyama, H.; Takemura, M.; Yasugi, Y.; Kishimoto, H.; Tamai, I.; Wang, J.; et al. Multiple Transport Mechanisms Involved in the Intestinal Absorption of Metformin: Impact on the Nonlinear Absorption Kinetics. J. Pharm. Sci. 2022, 111, 1531–1541.
- Zhou, M.; Xia, L.; Wang, J. Metformin Transport by a Newly Cloned Proton-Stimulated Organic Cation Transporter (Plasma Membrane Monoamine Transporter) Expressed in Human Intestine. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 1956–1962.
- Chen, E.C.; Liang, X.; Yee, S.W.; Geier, E.G.; Stocker, S.L.; Chen, L.; Giacomini, K.M. Targeted Disruption of Organic Cation Transporter 3 Attenuates the Pharmacologic Response to Metformin. Mol. Pharmacol. 2015, 88, 75–83.
- Müller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.H.; Koepsell, H.; Brandsch, M. Drug Specificity and Intestinal Membrane Localization of Human Organic Cation Transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860.
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways: Pharmacokinetics and Pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 820–827.
- Jin, H.-E.; Hong, S.-S.; Choi, M.-K.; Maeng, H.-J.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Reduced Antidiabetic Effect of Metformin and Down-Regulation of Hepatic Oct1 in Rats with Ethynylestradiol-Induced Cholestasis. Pharm. Res. 2009, 26, 549–559.
- Shu, Y.; Brown, C.; Castro, R.A.; Shi, R.J.; Lin, E.T.; Owen, R.P.; Sheardown, S.A.; Yue, L.; Burchard, E.G.; Brett, C.M.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1, OCT1, on Metformin Pharmacokinetics. Clin. Pharmacol. Ther. 2008, 83, 273–280.
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K.-I. Metformin Is a Superior Substrate for Renal Organic Cation Transporter OCT2 Rather than Hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386.
- Wilcock, C.; Bailey, C.J. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica Fate Foreign Compd. Biol. Syst. 1994, 24, 49–57.
- Shingaki, T.; Hume, W.E.; Takashima, T.; Katayama, Y.; Okauchi, T.; Hayashinaka, E.; Wada, Y.; Cui, Y.; Kusuhara, H.; Sugiyama, Y.; et al. Quantitative Evaluation of MMate1 Function Based on Minimally Invasive Measurement of Tissue Concentration Using PET with Metformin in Mouse. Pharm. Res. 2015, 32, 2538–2547.
- Tucker, G.T.; Casey, C.; Phillips, P.J.; Connor, H.; Ward, J.D.; Woods, H.F. Metformin Kinetics in Healthy Subjects and in Patients with Diabetes Mellitus. Br. J. Clin. Pharmacol. 1981, 12, 235–246.
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162.
- Choi, M.-K.; Jin, Q.-R.; Jin, H.-E.; Shim, C.-K.; Cho, D.-Y.; Shin, J.-G.; Song, I.-S. Effects of Tetraalkylammonium Compounds with Different Affinities for Organic Cation Transporters on the Pharmacokinetics of Metformin. Biopharm. Drug Dispos. 2007, 28, 501–510.
- Sato, T.; Masuda, S.; Yonezawa, A.; Tanihara, Y.; Katsura, T.; Inui, K.-I. Transcellular Transport of Organic Cations in Double-Transfected MDCK Cells Expressing Human Organic Cation Transporters HOCT1/HMATE1 and HOCT2/HMATE1. Biochem. Pharmacol. 2008, 76, 894–903.
- Chen, Y.; Teranishi, K.; Li, S.; Yee, S.W.; Hesselson, S.; Stryke, D.; Johns, S.J.; Ferrin, T.E.; Kwok, P.; Giacomini, K.M. Genetic Variants in Multidrug and Toxic Compound Extrusion-1, HMATE1, Alter Transport Function. Pharm. J. 2009, 9, 127–136.
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and Functional Characterization of a New Human Kidney-Specific H+/Organic Cation Antiporter, Kidney-Specific Multidrug and Toxin Extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135.
- Gan, S.C.; Barr, J.; Arieff, A.I.; Pearl, R.G. Biguanide-Associated Lactic Acidosis. Case Report and Review of the Literature. Arch. Intern. Med. 1992, 152, 2333–2336.
- Wang, D.; De Vivo, D. Pyruvate Carboxylase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Soeters, P.B.; Shenkin, A.; Sobotka, L.; Soeters, M.R.; de Leeuw, P.W.; Wolfe, R.R. The Anabolic Role of the Warburg, Cori-Cycle and Crabtree Effects in Health and Disease. Clin. Nutr. Edinb. Scotl. 2021, 40, 2988–2998.
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory Metabolism: Glycolysis, the TCA Cycle and Mitochondrial Electron Transport. Curr. Opin. Plant Biol. 2004, 7, 254–261.
- Rajasurya, V.; Anjum, H.; Surani, S. Metformin Use and Metformin-Associated Lactic Acidosis in Intensive Care Unit Patients with Diabetes. Cureus 2019, 11, e4739.
- Jeppesen, J.; Zhou, M.Y.; Chen, Y.D.; Reaven, G.M. Effect of Metformin on Postprandial Lipemia in Patients with Fairly to Poorly Controlled NIDDM. Diabetes Care 1994, 17, 1093–1099.
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5.
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel Gut-Based Pharmacology of Metformin in Patients with Type 2 Diabetes Mellitus. PLoS ONE 2014, 9, e100778.
- Mannucci, E.; Tesi, F.; Bardini, G.; Ognibene, A.; Petracca, M.G.; Ciani, S.; Pezzatini, A.; Brogi, M.; Dicembrini, I.; Cremasco, F.; et al. Effects of Metformin on Glucagon-like Peptide-1 Levels in Obese Patients with and without Type 2 Diabetes. Diabetes Nutr. Metab. 2004, 17, 336–342.
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K.T. Metformin Activates a Duodenal Ampk-Dependent Pathway to Lower Hepatic Glucose Production in Rats. Nat. Med. 2015, 21, 506–511.
- Grosskopf, I.; Ringel, Y.; Charach, G.; Maharshak, N.; Mor, R.; Iaina, A.; Weintraub, M. Metformin Enhances Clearance of Chylomicrons and Chylomicron Remnants in Nondiabetic Mildly Overweight Glucose-Intolerant Subjects. Diabetes Care 1997, 20, 1598–1602.
- Karise, I.; Bargut, T.C.; Del Sol, M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Metformin Enhances Mitochondrial Biogenesis and Thermogenesis in Brown Adipocytes of Mice. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 111, 1156–1165.
- Virtanen, K.A.; Hällsten, K.; Parkkola, R.; Janatuinen, T.; Lönnqvist, F.; Viljanen, T.; Rönnemaa, T.; Knuuti, J.; Huupponen, R.; Lönnroth, P.; et al. Differential Effects of Rosiglitazone and Metformin on Adipose Tissue Distribution and Glucose Uptake in Type 2 Diabetic Subjects. Diabetes 2003, 52, 283–290.
- Kim, E.K.; Lee, S.H.; Jhun, J.Y.; Byun, J.K.; Jeong, J.H.; Lee, S.-Y.; Kim, J.K.; Choi, J.Y.; Cho, M.-L. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing FGF21. Mediators Inflamm. 2016, 2016, 5813030.
- Wang, C.; Liu, F.; Yuan, Y.; Wu, J.; Wang, H.; Zhang, L.; Hu, P.; Li, Z.; Li, Q.; Ye, J. Metformin Suppresses Lipid Accumulation in Skeletal Muscle by Promoting Fatty Acid Oxidation. Clin. Lab. 2014, 60, 887–896.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
08 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No