Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MAYUR VIRARKAR | -- | 2570 | 2022-07-05 19:53:54 | | | |
| 2 | Conner Chen | Meta information modification | 2570 | 2022-07-06 05:11:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Virarkar, M.; Vulasala, S.S.; Gopireddy, D.; Morani, A.C.; Daoud, T.; Waters, R.; Bhosale, P. Renal Primary Neuroendocrine Neoplasms. Encyclopedia. Available online: https://encyclopedia.pub/entry/24844 (accessed on 27 July 2026).
Virarkar M, Vulasala SS, Gopireddy D, Morani AC, Daoud T, Waters R, et al. Renal Primary Neuroendocrine Neoplasms. Encyclopedia. Available at: https://encyclopedia.pub/entry/24844. Accessed July 27, 2026.
Virarkar, Mayur, Sai Swarupa Vulasala, Dheeraj Gopireddy, Ajaykumar C. Morani, Taher Daoud, Rebecca Waters, Priya Bhosale. "Renal Primary Neuroendocrine Neoplasms" Encyclopedia, https://encyclopedia.pub/entry/24844 (accessed July 27, 2026).
Virarkar, M., Vulasala, S.S., Gopireddy, D., Morani, A.C., Daoud, T., Waters, R., & Bhosale, P. (2022, July 05). Renal Primary Neuroendocrine Neoplasms. In Encyclopedia. https://encyclopedia.pub/entry/24844
Virarkar, Mayur, et al. "Renal Primary Neuroendocrine Neoplasms." Encyclopedia. Web. 05 July, 2022.
Copy Citation
Primary renal neuroendocrine neoplasms (NENs) are rare neoplasms. The 2016 WHO classification of renal tumors classified renal neuroendocrine neoplasms into well-differentiated NET, small-cell NEC, and large-cell NEC
neuroendocrine tumors
renal
1. Epidemiology, Presentation, and Pathogenesis
Primary renal NENs are rare neoplasms with around 100 cases reported in the literature, as per our knowledge (Figure 1) [1]. The 2016 WHO classification of renal tumors classified renal neuroendocrine neoplasms into well-differentiated NET, small-cell NEC, and large-cell NEC (Table 1) [2]. Renal carcinoid tumors, also known as well-differentiated NET of the kidneys, are rare, slow-growing neoplasms. They were first described by Resnick et al. in 1966 [3][4]. In a study by Hansel et al., the median age of presentation for renal NENs is 52 years, and the tumor equally affected male and female patients [5]. Primary renal carcinoid is the second most common genitourinary carcinoid tumor after testicular/ovarian carcinoids [6]. It is asymptomatic and incidentally detected in 25–30% of patients [4][7]. Typical symptoms of the carcinoid syndrome, including flushing, edema, and diarrhea, are uncommon and are seen in <10–15% of patients [3][8][9][10]. The incidence of small-cell renal NEC is rare; approximately 50 cases have been reported in the literature [11][12][13][14][15]. Most of the patients present at the age of 59 years with non-specific symptoms, such as hematuria, weight loss, and abdominal pain. There is no gender predilection, and the right kidney is more commonly involved than the left, with a ratio of 1.5:1 [11]. Evaluation with biochemical tests such as urinary–HIAA and serum chromogranin A and imaging is recommended to assess suspected NENs [16]. Death is seen in around 75% of patients within a year of diagnosis. Primary renal large cell NEC is an extremely rare malignancy; fewer than 10 cases have been reported in the literature [12][17][18][19][20][21][22]. Renal paragangliomas are rare, and to date, very few cases have been reported in the literature [23].
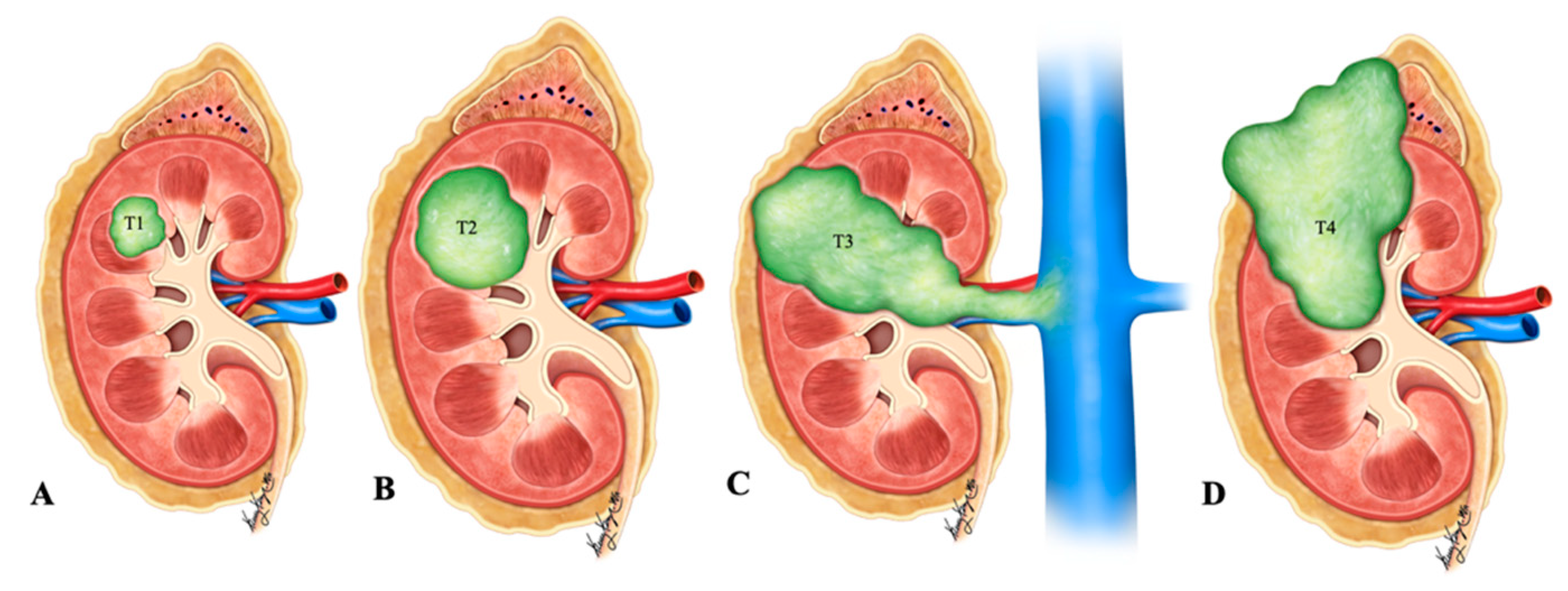
Figure 1. Illustration of renal neuroendocrine carcinoma. (A) T1: tumor limited to kidney and ≤7 cm in greatest dimension; (B) T2: tumor limited to kidney and >7 cm in dimension; (C) T3: tumor extending into the major vein or the tissue around the kidney; (D) T4: tumor growth beyond the Gerota’s fascia and may be growing into the adrenal gland.
| Renal Carcinoids | Renal Small-Cell NEC | Renal Large-Cell NEC | Renal Paraganglioma | |
|---|---|---|---|---|
| Cellular arrangements | Trabecular/gyriform, glandular, insular, solid or mixed patterns | Sheets, nests, trabeculae | Solid nesting growth pattern | |
| Cellular characteristics | Round or polygonal cells with granular cytoplasm. | Poorly differentiated small round to fusiform cells; scanty stroma and cytoplasm | Large cells with abundant cytoplasm | Round to oval cells (Zellballen); sometimes spindle-shaped elongated cells; abundant granular cytoplasm; |
| Nuclei | Round nucleus with finely stippled ribbon-like chromatin; inconspicuous nucleoli | Hyperchromatic nuclei; Nuclear molding; stippled/dispersed chromatin; inconspicuous nucleoli | Pleomorphic nuclei with vesicular chromatin; Prominent nucleoli | Variant nuclei with regular uniform chromatin or hyperchromasia |
| Additional features | Calcifications (25% of cases); Absence of frequent mitosis; Low Ki-67 proliferation index | Extensive tumor necrosis; perivascular DNA deposition (Azzopardi phenomenon); Brisk mitoses; Calcifications can be seen. | High mitotic rate (>10/10 per HPF); Large necrotic areas | Highly vascular intervening stroma; Low mitotic index and necrosis |
| Immunohistochemistry | Positive for cytokeratin, chromogranin, synaptophysin, gremileus and neuron-specific enolase | Dot-like cytokeratin staining in the cytoplasm; Variable positivity for chromogranin A, synaptophysin, CD56 and NSE; A few cases reported positive staining for TTF-1 [15][29][30] | Diffusely and strongly positive for synaptophysin | Positive for INSM1, synaptophysin and chromogranin; Negative for keratins; Positive tyrosine hydroxylase and nuclear GATS3; scattered S100-sustentacular cells |
2. Pathogenesis
There are many hypotheses explaining the cellular origins of neuroendocrine neoplasms in the kidneys: tumors (i) arise from renal stem cells that progress towards neuroendocrine lineage [16] or (ii) arise from entrapped neural crest cells in the kidney during embryogenesis [31] or (iii) develop alongside congenital abnormalities of the kidney [16] or (iv) are secondary to the metaplasia of pyelocaliceal urothelium resulting from chronic inflammation [24]. Renal NENs arises most commonly in individuals with congenital or acquired renal anomalies, such as horseshoe kidney (26% of renal NENs), polycystic disease, and mature cystic teratoma (15% of renal NENs) [32][33][34][35][36]. Krishnan et al. speculated that renal NENs arises from the already existing hyperplasia of neuroendocrine cells in the metastatic or teratomatous epithelium of the horseshoe kidney [32]. Primary renal small-cell NEC is a poorly differentiated tumor with a poor prognosis that is similar to small-cell NEC at other sites. Unlike renal parenchymal small-cell NEC, the ones arising from the renal pelvis are associated with urothelial or squamous cell carcinoma [15][37][38]. The origin of renal small-cell NEC is different from that of clear cell renal cell carcinoma. The pattern of chromosomal aberration can explain this. Gain of multiple chromosomes, loss of p53, and amplification of MYC are seen in small-cell renal NEC, whereas loss of the short arm of chromosome 3 (3p) is observed in clear cell renal cell carcinoma [26]. However, El-Naggar reported a loss of heterozygosity in chromosome 3p21 and suggested that this event is preliminary to all the renal neoplasms [39]. Renal paragangliomas are usually benign, and around 10% of patients encounter malignant transformation [27]. Classic cancer predisposition syndromes, including von Hippel–Lindau, multiple endocrine neoplasia type-2, and neurofibromatosis type 1 disease are associated with paragangliomas. Metastases are rare in VHL and MEN2 associated paragangliomas, whereas they may occur in 12% of NF-1 associated diseases [40]. The risk of metastases is high in succinate dehydrogenase subunits B (25–50%) and A (12%) [41].
3. Imaging
Ultrasound of renal NENs shows an iso-to hyperechoic mass with a peripheral anechoic or hypoechoic rim [9][10][42]. Computed tomography demonstrates a well-circumscribed solid, hypodense tumor with minimal enhancement in contrast-enhanced venous phase CT that corresponds to hypo- or avascular lesions on angiography (Figure 2, Figure 3 and Figure 4) [16][43]. Heterogeneity and calcifications on CT can be seen in 60% and 26% of cases [7][31]. Around 75% of patients present with tumors > 4 cm, and >50% of patients demonstrate extrarenal invasion (perinephric, renal vein, or renal sinus fat) or distant spread of tumor [10][44]. However, differentiation is challenging, solely based on CT or MR imaging findings among renal NENs and renal cell carcinoma (Table 2). In addition, smaller carcinoids are difficult to visualize on conventional imaging. Octreotide scintigraphy complements CT or MRI and aids in diagnostic evaluation, staging, and follow-up of tumors and metastases with high sensitivity (85%) [11][45]. However, the major limitation is the uptake of tracer material by the normal renal parenchyma, obscuring the suspicious lesion.
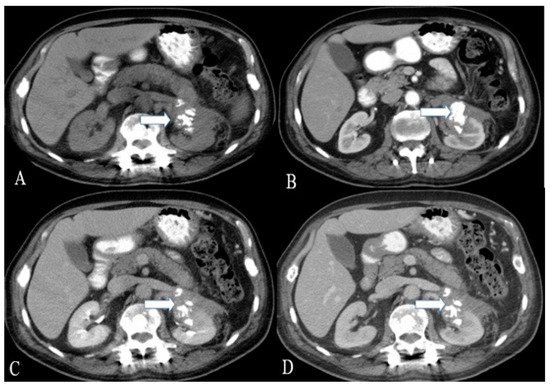
Figure 2. A 36-year-old female with renal NENs. CT of the abdomen before (A) and after oral and IV contrast administration in (B) arterial, (C) porto-venous, and (D) delayed phases reveal a 5.3 × 3.9 cm partially calcified mass (arrows) arising from the left kidney, extending to the left perinephric space, and abutting the tail of the pancreas. Pathology revealed small-cell carcinoma with neuroendocrine differentiation.
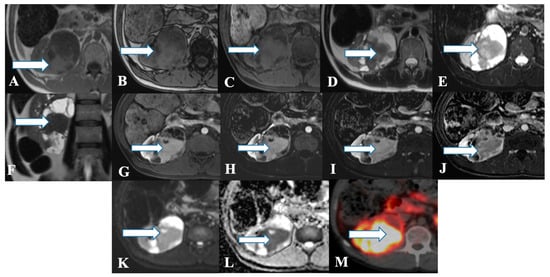
Figure 3. A 45-year-old female with renal NENs. MRI axial T1 (A) in phase, (B) out of phase, (C) 3D axial, (D) T2, (E) T2 fat saturation, (F) coronal T2, (G) post-contrast dynamic, (H,I) dynamic subtraction images, (J) delayed postcontrast axial, (K) DWI, and (L) ADC show a large right renal mid and lower pole mass (arrows) measuring 6.6 × 7.1 cm showing central enhancing solid component with restricted diffusion and peripheral cystic component. Pathology revealed a neuroendocrine tumor. (M) Axial fused PET/CT images using Gallium 68 Dotatate show heterogeneously avid DOTATATE uptake of the mass (arrow), in keeping with biopsy-confirmed right kidney neuroendocrine carcinoma.
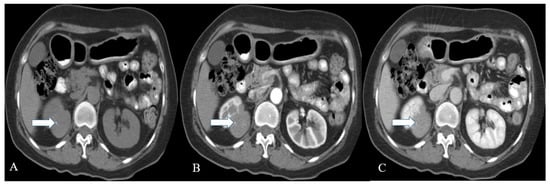
Figure 4. A 57-year-old female with renal NENs. CT scan of the abdomen before (A) and after oral and IV contrast administration in arterial (B) and porto-venous (C) phases reveal a right renal upper polar well-defined mass (arrow) with flecks of calcifications, measuring 5 × 4 cm encroaching on the right perinephric space without appreciable involvement of the right renal hilum. Pathology revealed well-differentiated neuroendocrine carcinoma (atypical carcinoid).
Table 2. Imaging features of solid renal masses [46].
| Renal Lesion | CT | MRI |
|---|---|---|
| Clear Cell carcinoma | Heterogeneous mass due to necrotic, cystic and hemorrhagic areas; Strong contrast enhancement in corticomedullary and contrast wash out during nephrographic phases | Hyper vascular lesion; Hyperintense on T2WI and hypo- to isointense on T1WI; Heterogeneous avid enhancement than the rest of RCC types on contrast administration; Microscopic fat in 60% of cases; CSI: >25% signal loss on opposed phase relative to in-phase imaging due to fat content; Tumor pseudo capsule: hypointense rim on T1WI and T2WI. |
| Chromophobe carcinoma | Homogeneous to heterogeneous mass | Well circumscribed homogeneous tumors; Iso- to hypointense lesion son T2WI; The lesion enhances greater than papillary but lesser than clear cell renal carcinoma; Central stellate scar in 30–40% of cases; Spoke-wheel enhancement can be observed; Segmental enhancement inversion may be noticed; Calcifications in 38% of cases. |
| Papillary carcinoma | Tumors < 3 cm: homogeneous; Tumors ≥ 4 cm: Heterogeneous due to necrosis; Subtle contrast enhancement than ccRCC; Absent enhancement can be observed in 25% of patients; | Well-circumscribed homogeneous mass; Usually <3 cm; Mass: hypointense on T2WI which enhance progressively with contrast administration; CSI: signal loss on in-phase relative to opposed phase imaging due to hemosiderin deposition; Fibrous capsule: hypointense on T1WI & T2WI; |
| Renal NET | Heterogeneous solid tumor with cystic component as well; Minimal enhancement on contrast administration; Octreotide scintigraphy: High affinity for somatostatin in 87% of patients. | Heterogeneous signal intensity on T1 and T2WI with areas of high signal intensity on T1WI due to hemorrhage; The mass enhances with contrast administration |
Renal paragangliomas are more common in the kidney’s upper pole, and all patients shall have a biochemical evaluation before surgical resection [47]. Patients with hereditary syndrome-associated paragangliomas are more likely to develop multifocal primary tumors with varying degrees of metastases [48]. Paragangliomas are well-defined tumors with hemorrhagic and necrotic areas on CT, and calcification in 15% of cases [49]. On contrast administration, they exhibit intense homogeneous enhancement secondary to the tumor hypervascularity [49]. Since the MR resolution of tumors is better than that of CT, MR imaging has higher sensitivity in detecting liver metastases and blood vessel invasion [50]. In MR imaging, paragangliomas demonstrate low to intermediate signal intensity on T1-weighted, signal voids on T1-weighted spin-echo, and high signal intensity on T2-weighted imaging sequences [49]. Functional imaging, iodine 123 metaiodobenzylguanidine (I123-MIBG), is widely used to identify metastases and differentiate functional from nonfunctional paragangliomas.
I123-MIBG has high specificity (95–100%) but a low detection rate; the sensitivity ranges from 56 to 72% [51][52][53][54]. The application of PET/CT with 68Ga-DOTA peptides and 18F-fluorodeoxyglucose has increased recently. PET/CT imaging with 68Ga-labeled somatostatin analogs such as DOTATOC, DOTATATE, and DOTANOC is being employed, and it targets the over-expressive somatostatin receptor subtype 2 in the paragangliomas [55]. DOTATATE PET/CT is the better imaging modality in patients with well-differentiated renal NENs. 18F-FDG PET/CT is superior to the 68Ga-based PET/CT, 123I-MIBG, CT, or MRI for high-grade NEC and detects the metastatic or recurrent tumors. It exhibits increased 18F-FDG uptake in SDH and VHL-related tumors and decreased uptake in MEN-2-related tumors. In addition, fluorine-18-labeled dihydroxyphenylalanine, a new tracer, was found to acquire images within hours; compare that to a day for an MIBG scan. The main limitation of FDG-PET is that its specificity is lower than that of 123I-MIBG and 111-In Octreoscan [56][57].
The diagnosis of NENs is based on histopathological and immunohistochemical characteristics of the tumor (Figure 5). Grossly, the renal NENs is solitary with well-delineated borders from the surrounding normal parenchyma and demonstrates solid (74% of cases) and cystic (49% of cases) surfaces, alongside focal calcification (30% of cases) and the occasional pseudo-capsule [45]. Usually, as the retroperitoneal space is highly distensible, the patients manifest with large tumor masses at diagnosis [31]. In their study, Hansel et al. described the average tumor size being from 2.6 to 17 cm. They reported that around 62% of patients demonstrated metastasis in regional lymph nodes or distant organs [1][5]. Hence, it can be said that the metastases are directly related to the tumor size in the renal NENs [1][58].
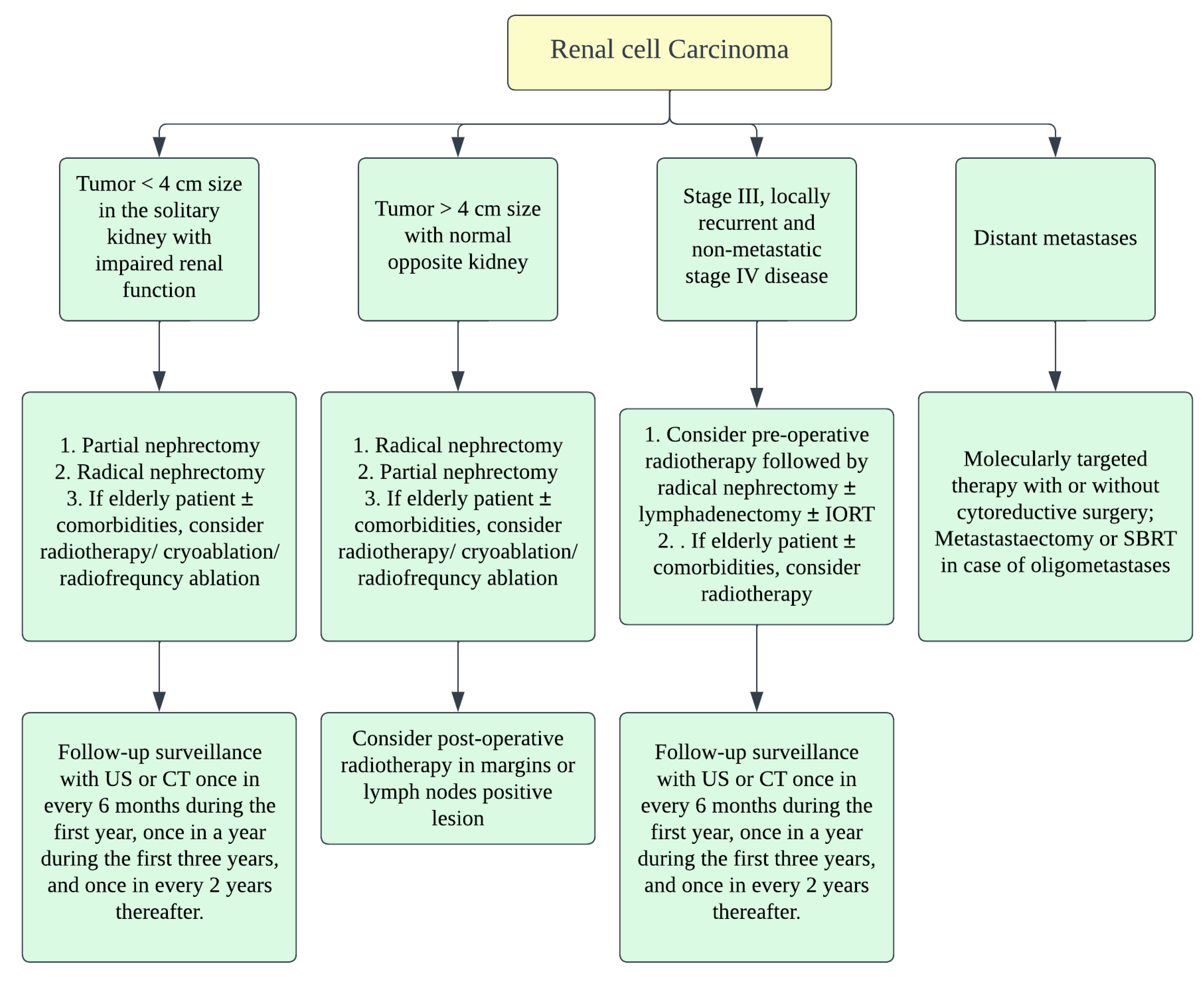
Figure 5. Treatment algorithm of renal malignancies.
4. Prognosis and Management
Renal NENs have higher survival rates than poorly differentiated renal NENs [1]. Age > 40 years, tumor size > 4 cm, extrarenal tumor invasion, purely solid surface, and >1 mitoses per HPF are the poor prognostic factors in patients with renal NENs [7][8][59]. In the presence of horseshoe kidneys, the carcinoids manifest indolent behavior [10][31][60]. Zhenglin et al. reported regional or distant metastases in 83% and a median overall survival of only 27 months in patients with renal NEC [1]. Patients with renal paraganglioma and metastases at diagnosis have 5-year survival rates ranging from 50 to 70% [28].
Surgery is the mainstay of treatment in renal NENs. The staging of renal NENs is described in Table 3 and Table 4. The standard treatment for localized primary renal NENs is nephrectomy with a lymph node dissection (Figure 6) [16]. As the surgery could be a prognostic factor, radical or partial nephrectomy is the treatment choice for localized or metastatic renal NENs [31]. Resection of the entire visible tumor is recommended in localized renal NEC. Rosenberg et al. described that, as carcinoids tend to be benign, partial nephrectomy or cryoablation of the tumor shall be the standard treatment; therefore, functional nephrons can be spared [61]. Chemotherapy and radiation have no proven benefit in managing NENs in general [11]. When indicated, open surgical resection shall be considered in the case of primary paraganglioma. Cytotoxic chemotherapy is recommended as the first-line therapy in patients with a high tumor burden (extensive metastases), symptoms, or rapid progression. Clinically used chemotherapeutic medications include CVD (cyclophosphamide/vincristine/dacarbazine) or temozolomide-based regimen. I131 MIBG therapy can be considered in patients requiring systemic treatment and who have MIBG-avid disease [28].
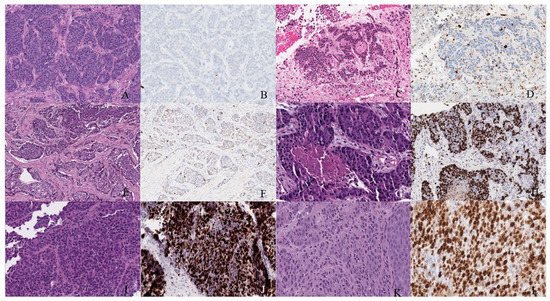
Figure 6. Well-differentiated NET (neuroendocrine tumor). (A) Grade 1 shows packets of neuroendocrine cells separated by fibrovascular tissue. (B) Ki67 immunohistochemical stain shows a proliferation rate of 1%. Grade 2 well-differentiated NET showing (C) trabecular architecture. (D) Ki67 immunohistochemical stain shows a proliferation rate of 15%. Grade 3 well-differentiated NET with a (E) solid and nested pattern. (F) Ki67 immunohistochemical stain shows a proliferation rate of 30%. Poorly differentiated neuroendocrine carcinoma. (G) The tumor shows solid nests of poorly differentiated epithelioid cells with elevated nuclear size, pleomorphism, and dense chromatin. Notice there is necrosis in the center of the image. (H) Ki67 immunohistochemical stain shows a proliferation rate of 80%. Small cell neuroendocrine carcinoma (I) sheets of small to medium, round/oval, blue cells with minimal cytoplasm. The chromatin is finely dispersed. (J) Nuclei demonstrate molding. Ki67 shows a proliferation index of 80%. Large cell neuroendocrine carcinoma (K) with organoid architecture. Large cells (~3× size of small-cell carcinoma) are present with abundant cytoplasm, variably coarse chromatin, nuclear pleomorphism, and prominent nucleoli. (L) Ki67 immunohistochemical stain shows a proliferation rate of 90%.
Table 3. AJCC staging of renal malignancies.
| Stage | TNM Category | Description |
|---|---|---|
| I | T1 N0 M0 | T1–Tumor limited to kidney & ≤7 cm in greatest dimension N0–No lymph node involvement M0–No distant metastasis |
| II | T2 N0 M0 | T2–Tumor limited to kidney & >7 cm in greatest dimension |
| III | T3 N0 M0 | T3–Tumor extending into the major vein or into the tissue around kidney; but not growing into adrenal gland or beyond Gerota’s fascia |
| T1-3 N1 M0 | N1–Tumor spread to regional lymph nodes | |
| IV | T4 Any N M0 | T4–Tumor beyond Gerota’s fascia and may be growing into adrenal gland on the top of kidney |
| Any T Any N M1 | M1–Distant lymph nodes and/or other organs. |
Table 4. AJCC staging of paragangliomas [28].
| Stage | TNM Category | Description |
|---|---|---|
| I | T1 N0 M0 | T1–Pheochromocytoma < 5 cm in greatest dimension; No extra-adrenal invasion; N0–no lymph node metastases; M0–No distant metastases |
| II | T2 N0 M0 | T2–Pheochromocytoma ≥ 5 cm, sympathetic paraganglioma of any size; no extra-adrenal invasion |
| III | T1 N1 M0 | N1–Lymph node metastases |
| III | T2 N1 M0 | |
| III | T3 N0 M0 | T3–Tumor of any size with surround tissue invasion (liver, spleen, pancreas and kidneys) |
| III | T3 N1 M0 | |
| IV | Any T Any N M1 | M1–distant metastases; M1a–Distant metastases to only bone; M1b–Distant metastases to only lymph nodes/liver/lung; M1c–Distant metastases to bone plus multiple other sites |
References
- Yi, Z.; Liu, R.; Hu, J.; He, T.; Wang, Z.; Li, Y.; Zu, X. Clinicopathologic Features and Survival Outcomes for Primary Renal Neuroendocrine Neoplasms. Clin. Genitourin. Cancer 2021, 19, 155–161.
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105.
- Resnick, M.E.; Unterberger, H.; McLoughlin, P.T. Renal carcinoid producing the carcinoid syndrome. Med. Times 1966, 94, 895–896.
- Murali, R.; Kneale, K.; Lalak, N.; Delprado, W. Carcinoid tumors of the urinary tract and prostate. Arch. Pathol. Lab. Med. 2006, 130, 1693–1706.
- Hansel, D.E.; Epstein, J.I.; Berbescu, E.; Fine, S.W.; Young, R.H.; Cheville, J.C. Renal carcinoid tumor: A clinicopathologic study of 21 cases. Am. J. Surg. Pathol. 2007, 31, 1539–1544.
- Armah, H.B.; Parwani, A.V.; Perepletchikov, A.M. Synchronous primary carcinoid tumor and primary adenocarcinoma arising within mature cystic teratoma of horseshoe kidney: A unique case report and review of the literature. Diagn. Pathol. 2009, 4, 17.
- Romero, F.R.; Rais-Bahrami, S.; Permpongkosol, S.; Fine, S.W.; Kohanim, S.; Jarrett, T.W. Primary carcinoid tumors of the kidney. J. Urol. 2006, 176, 2359–2366.
- Raslan, W.F.; Ro, J.Y.; Ordonez, N.G.; Amin, M.B.; Troncoso, P.; Sella, A.; Ayala, A.G. Primary carcinoid of the kidney. Immunohistochemical and ultrastructural studies of five patients. Cancer 1993, 72, 2660–2666.
- Moulopoulos, A.; DuBrow, R.; David, C.; Dimopoulos, M.A. Primary renal carcinoid: Computed tomography, ultrasound, and angiographic findings. J. Comput. Assist. Tomogr. 1991, 15, 323–325.
- Katabathina, V.S.; Vikram, R.; Olaoya, A.; Paspulati, R.M.; Nicolas, M.M.; Rao, P.; Zaheer, A.; Prasad, S.R. Neuroendocrine neoplasms of the genitourinary tract in adults: Cross-sectional imaging spectrum. Abdom. Radiol. 2017, 42, 1472–1484.
- Posfai, B.; Kuthi, L.; Varga, L.; Laczo, I.; Revesz, J.; Kranicz, R.; Maraz, A. The Colorful Palette of Neuroendocrine Neoplasms in the Genitourinary Tract. Anticancer Res. 2018, 38, 3243–3254.
- Lane, B.R.; Chery, F.; Jour, G.; Sercia, L.; Magi-Galluzzi, C.; Novick, A.C.; Zhou, M. Renal neuroendocrine tumours: A clinicopathological study. BJU Int. 2007, 100, 1030–1035.
- González-Lois, C.; Madero, S.; Redondo, P.; Alonso, I.; Salas, A.; Montalbán, M.A. Small cell carcinoma of the kidney: A case report and review of the literature. Arch. Pathol. Lab. Med. 2001, 125, 796–798.
- Akkaya, B.K.; Mustafa, U.; Esin, O.; Turker, K.; Gulten, K. Primary small cell carcinoma of the kidney. Urol. Oncol. Semin. Orig. Investig. 2003, 21, 11–13.
- Si, Q.; Dancer, J.; Stanton, M.L.; Tamboli, P.; Ro, J.Y.; Czerniak, B.A.; Shen, S.S.; Guo, C.C. Small cell carcinoma of the kidney: A clinicopathologic study of 14 cases. Hum. Pathol. 2011, 42, 1792–1798.
- Nguyen, A.H.; O’Leary, M.P.; De Andrade, J.P.; Ituarte, P.H.G.; Kessler, J.; Li, D.; Singh, G.; Chang, S. Natural History of Renal Neuroendocrine Neoplasms: A NET by Any Other Name? Front. Endocrinol. 2020, 11, 624251.
- Ratnagiri, R.; Singh, S.S.; Majhi, U. Large-cell neuroendocrine carcinoma of the kidney: Clinicopathologic features. Indian J. Urol. IJU J. Urol. Soc. India 2009, 25, 274.
- Dundr, P.; Pešl, M.; Povýšil, C.; Bauerová, L.; Soukup, V. Primary large cell neuroendocrine carcinoma of the kidney. Pathol. Oncol. Res. 2010, 16, 139–142.
- Palumbo, C.; Talso, M.; Dell’Orto, P.G.; Cozzi, G.; De Lorenzis, E.; Conti, A.; Maggioni, M.; Cesare Rocco, B.M.; Maggioni, A.; Rocco, F. Primary large cell neuroendocrine carcinoma of the renal pelvis: A case report. Urol. J. 2014, 81, 57–59.
- Wann, C.; John, N.T.; Kumar, R.M. Primary renal large cell neuroendocrine carcinoma in a young man. J. Clin. Diagn. Res. JCDR 2014, 8, ND08.
- Shimbori, M.; Osaka, K.; Kawahara, T.; Kasahara, R.; Kawabata, S.; Makiyama, K.; Kondo, K.; Nakaigawa, N.; Yamanaka, S.; Yao, M. Large cell neuroendocrine carcinoma of the kidney with cardiac metastasis: A case report. J. Med. Case Rep. 2017, 11, 297.
- Sanguedolce, F.; Calo, B.; Chirico, M.; Tortorella, S.; Carrieri, G.; Cormio, L. Urinary Tract Large Cell Neuroendocrine Carcinoma: Diagnostic, Prognostic and Therapeutic Issues. Anticancer Res. 2020, 40, 2439–2447.
- Yi, C.; Han, L.; Yang, R.; Yu, J. Paraganglioma of the renal pelvis: A case report and review of literature. Tumori 2017, 103, e47–e49.
- Lamb, L.; Shaban, W. Primary renal carcinoid tumor: A radiologic review. Radiol. Case Rep. 2014, 9, 923.
- Mazzucchelli, R.; Morichetti, D.; Lopez-Beltran, A.; Cheng, L.; Scarpelli, M.; Kirkali, Z.; Montironi, R. Neuroendocrine tumours of the urinary system and male genital organs: Clinical significance. BJU Int. 2009, 103, 1464–1470.
- Shehabeldin, A.N.; Ro, J.Y. Neuroendocrine tumors of genitourinary tract: Recent advances. Ann. Diagn. Pathol. 2019, 42, 48–58.
- Bahar, B.; Pambuccian, S.E.; Gupta, G.N.; Barkan, G.A. Renal paraganglioma: Report of a case managed by robotic assisted laparoscopic partial nephrectomy and review of the literature. Case Rep. Urol. 2014, 2014, 527592.
- Fishbein, L.; Del Rivero, J.; Else, T.; Howe, J.R.; Asa, S.L.; Cohen, D.L.; Dahia, P.L.; Fraker, D.L.; Goodman, K.A.; Hope, T.A. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021, 50, 469–493.
- Kuroda, N.; Imamura, Y.; Hamashima, T.; Ohe, C.; Mikami, S.; Nagashima, Y.; Inoue, K.; Perez-Montiel, D.; Petersson, F.; Michal, M.; et al. Review of small cell carcinoma of the kidney with focus on clinical and pathobiological aspects. Pol. J. Pathol. 2014, 65, 15–19.
- Agoff, S.N.; Lamps, L.W.; Philip, A.T.; Amin, M.B.; Schmidt, R.A.; True, L.D.; Folpe, A.L. Thyroid transcription factor-1 is expressed in extrapulmonary small cell carcinomas but not in other extrapulmonary neuroendocrine tumors. Mod. Pathol. 2000, 13, 238–242.
- Korkmaz, T.; Seber, S.; Yavuzer, D.; Gumus, M.; Turhal, N.S. Primary renal carcinoid: Treatment and prognosis. Crit. Rev. Oncol. Hematol. 2013, 87, 256–264.
- Krishnan, B.; Truong, L.D.; Saleh, G.; Sirbasku, D.M.; Slawin, K.M. Horseshoe kidney is associated with an increased relative risk of primary renal carcinoid tumor. J. Urol. 1997, 157, 2059–2066.
- Takeshima, Y.; Inai, K.; Yoneda, K. Primary carcinoid tumor of the kidney with special reference to its histogenesis. Pathol. Int. 1996, 46, 894–900.
- Shibata, R.; Okita, H.; Shimoda, M.; Asakura, H.; Murai, M.; Sakamoto, M.; Hata, J. Primary carcinoid tumor in a polycystic kidney. Pathol. Int. 2003, 53, 317–322.
- De Hoog, J.P.; Murray, S.; Chou, W. Horseshoe kidney and primary renal carcinoid tumour: A case report of a rare entity. Grand Rounds 2010, 10, 46–51.
- Litwinowicz, R.; Szpor, J.; Januś, G.; Worek, M.; Okoń, K. Primary carcinoid tumour in horseshoe kidney. Pol. J. Pathol. 2011, 62, 72–74.
- Jeung, J.A.; Cao, D.; Selli, B.W.; Clapp, W.L.; Oliai, B.R.; Parwani, A.V.; Allan, R.W. Primary renal carcinoid tumors: Clinicopathologic features of 9 cases with emphasis on novel immunohistochemical findings. Hum. Pathol. 2011, 42, 1554–1561.
- Wang, X.; Wang, F.; Liang, Y.; Chen, W. Primary small cell carcinoma after renal transplant: A case report. Medicine 2018, 97, e12592.
- El-Naggar, A.K.; Troncoso, P.; Ordonez, N.G. Primary renal carcinoid tumor with molecular abnormality characteristic of conventional renal cell neoplasms. Diagn. Mol. Pathol. Am. J. Surg. Pathol. Part B 1995, 4, 48–53.
- Gruber, L.M.; Erickson, D.; Babovic-Vuksanovic, D.; Thompson, G.B.; Young, W.F., Jr.; Bancos, I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin. Endocrinol. 2017, 86, 141–149.
- Van Der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.; Corssmit, E.P.; Dinjens, W.N.; Van De Horst-schrivers, A.N.; Jansen, J.C.; De Jong, M.M.; Kunst, H.P.; Kusters, B. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: A nationwide study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445.
- McKeown, D.K.; Nguyen, G.-K.; Rudrick, B.; Johnson, M.A. Carcinoid of the kidney: Radiologic findings. Am. J. Roentgenol. 1988, 150, 143–144.
- Karaosmanoğlu, A.D.; Onur, M.R.; Shirkhoda, A.; Ozmen, M.; Hahn, P.F. Unusual Malignant Solid Neoplasms of the Kidney: Cross-Sectional Imaging Findings. Korean J. Radiol. 2015, 16, 853–859.
- Fine, S.W. Neuroendocrine lesions of the genitourinary tract. Adv. Anat. Pathol. 2007, 14, 286–296.
- Shurtleff, B.T.; Shvarts, O.; Rajfer, J. Carcinoid tumor of the kidney: Case report and review of the literature. Rev. Urol. 2005, 7, 229.
- Van Oostenbrugge, T.J.; Fütterer, J.J.; Mulders, P.F. Diagnostic imaging for solid renal tumors: A pictorial review. Kidney Cancer 2018, 2, 79–93.
- Lenders, J.W.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942.
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Rajiah, P.; Sinha, R.; Cuevas, C.; Dubinsky, T.J.; Bush, W.H., Jr.; Kolokythas, O. Imaging of uncommon retroperitoneal masses. Radiographics 2011, 31, 949–976.
- Dromain, C.; de Baere, T.; Baudin, E.; Galline, J.; Ducreux, M.; Boige, V.; Duvillard, P.; Laplanche, A.; Caillet, H.; Lasser, P. MR imaging of hepatic metastases caused by neuroendocrine tumors: Comparing four techniques. Am. J. Roentgenol. 2003, 180, 121–128.
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Havekes, B.; Eisenhofer, G.; Martiniova, L.; Adams, K.T.; Pacak, K. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2009, 94, 4757–4767.
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Eisenhofer, G.; King, K.S.; Rao, J.U.; Wesley, R.A.; Adams, K.T. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J. Natl. Cancer Inst. 2012, 104, 700–708.
- Jiang, Y.; Hou, G.; Zhu, Z.; Huo, L.; Cheng, W.; Li, F. The value of multiparameter (18)F-FDG PET/CT imaging in differentiating retroperitoneal paragangliomas from unicentric Castleman disease. Sci. Rep. 2020, 10, 12887.
- Lee, K.Y.; Oh, Y.W.; Noh, H.J.; Lee, Y.J.; Yong, H.S.; Kang, E.Y.; Kim, K.A.; Lee, N.J. Extraadrenal paragangliomas of the body: Imaging features. AJR Am. J. Roentgenol. 2006, 187, 492–504.
- Taïeb, D.; Pacak, K. Molecular imaging and theranostic approaches in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018, 372, 393–401.
- Katabathina, V.S.; Rajebi, H.; Chen, M.; Restrepo, C.S.; Salman, U.; Vikram, R.; Menias, C.O.; Prasad, S.R. Genetics and imaging of pheochromocytomas and paragangliomas: Current update. Abdom. Radiol. 2020, 45, 928–944.
- Castinetti, F.; Kroiss, A.; Kumar, R.; Pacak, K.; Taieb, D. 15 YEARS OF PARAGANGLIOMA: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2015, 22, T135-145.
- Omiyale, A.O.; Venyo, A.K. Primary carcinoid tumour of the kidney: A review of the literature. Adv. Urol. 2013, 2013, 579396.
- McGarrah, P.W.; Westin, G.F.M.; Hobday, T.J.; Scales, J.A.; Ingimarsson, J.P.; Leibovich, B.C.; Halfdanarson, T.R. Renal Neuroendocrine Neoplasms: A Single-center Experience. Clin. Genitourin. Cancer 2020, 18, e343–e349.
- Zhang, Q.; Ming, J.; Zhang, S.; Qiu, X. Primary micro neuroendocrine tumor arising in a horseshoe kidney with cyst: Report of a case and review of literature. Diagn. Pathol. 2012, 7, 126.
- Rosenberg, J.E.; Albersheim, J.A.; Sathianathen, N.J.; Murugan, P.; Weight, C.J. Five new cases of primary renal carcinoid tumor: Case reports and literature review. Pathol. Oncol. Res. 2020, 26, 341–346.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
842
Revisions:
2 times
(View History)
Update Date:
07 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No