 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dennis Kobelt | + 3655 word(s) | 3655 | 2020-09-25 08:38:54 | | | |
| 2 | Bruce Ren | Meta information modification | 3655 | 2020-10-10 09:56:46 | | |
Video Upload Options
Metastasis represents the most lethal attribute of cancer and critically limits successful therapies in many tumor entities. The clinical need is defined by the fact that all cancer patients, who have or who will develop distant metastasis, will experience shorter survival. Thus, the ultimate goal in cancer therapy is the restriction of solid cancer metastasis by novel molecularly targeted small molecule based therapies. Biomarkers identifying cancer patients at high risk for metastasis and simultaneously acting as key drivers for metastasis are extremely desired. Clinical interventions targeting these key molecules will result in high efficiency in metastasis intervention. In result of this, personalized tailored interventions for restriction and prevention of cancer progression and metastasis will improve patient survival.
1. Introduction
1.1. The Demanding Clinical Need for Metastasis Intervention
Despite the progress for treatment of solid cancers, metastasis remains the key issue impacting failure or success of cancer therapies. Metastatic dissemination of primary tumors is directly linked to patient survival. Metastasis is not an inherent property of all neoplastic cells [1]. Some cancers are highly aggressive forming metastases with high frequency, while others are rarely metastatic despite being locally invasive. But, metastasis is the most lethal attribute for cancer patients and counts for about 90% of all cancer deaths [2][3].
Further, metastatic spread critically limits successful therapies in many tumor entities [4]. The limited therapeutic success defines the clinical need for novel metastasis-inhibiting treatment strategies aiming at key events and drivers of metastasis formation by using small molecule drugs. We are focusing here on biomarkers acting as causal key drivers for metastasis, being involved in signaling pathways, promoting and driving the metastatic phenotype of cancer cells, which may serve as useful targets for small molecule-based restriction of metastasis formation.
1.2. Exploiting the Metastatic Cascade to Find Vulnerabilities for Metastasis Intervention
Here we dissect the metastatic cascade for novel approaches to combat metastasis formation, which arise upon reviewing the metastatic cascade [5][6]. The main steps of this cascade start with cellular transformation and tumor growth. This necessarily includes progressive growth of neoplastic cells and the availability of nutrients for the expanding tumor mass, initially supplied by simple diffusion. The second step is proliferation and angiogenesis. Here, the extensive vascularization must occur if a tumor mass is to exceed 1–2 mm in diameter. Angiogenic factors must be synthesized and secreted, thereby building a capillary network from the surrounding host tissue. The third step is detachment and invasion. Tumor cell detachment from the primary tumor mass is caused by loss of adhesion programs and invasion in the adjacent tissue is mainly characterized by degradation of the matrix using a variety of proteinases, both leading to increase in cell motility. This local invasion of the tumor cells into the host stroma paves the way of the detached and invasive tumor cell into circulation. The next step—intravasation, when tumor cells enter the blood vessel and circulation—is performed by single tumor cells or tumor cell aggregates. Although the majority of these circulating tumor cells are rapidly destroyed, some cells survive the circulation, staying dormant and are trapped in the capillary beds of distant organs. In the circulation, tumor cells interact with for example, platelets and lymphocytes. Then, circulating tumor cells arrest at distant organ sites by binding the endothelium of the vessels there [7]. During the extravasation step, educated tumor cells leave the circulation by rupture of the walls surrounding the vessel and penetration of the circulating tumor cells into adjacent tissue. The last step, completing metastasis formation, is the proliferation and the re-organization of the extracellular matrix (ECM) of the arrested tumor cells in the organs of the secondary site, essentially supported by an appropriate microenvironment. A newly generated vascular network of the micrometastases will help to evade destruction by host defenses. Metastases then grow into metastatic colonies, with about 50 cells will constitute a colony and continue to grow until macroscopic metastases are clinically detectable.
Thus, metastasis development is only possible when the “seed,” the tumor cells as the secondary site and the “soil,” the new surrounding organ, are compatible—the “seed and soil model” [8]. Further, since each of the steps of the metastatic cascade is dependent on clearly defined molecular pathways and networks, key targets of these signaling cascades can be identified and used for step-specific treatment [9]. Various interference opportunities have been developed using small molecules [10].
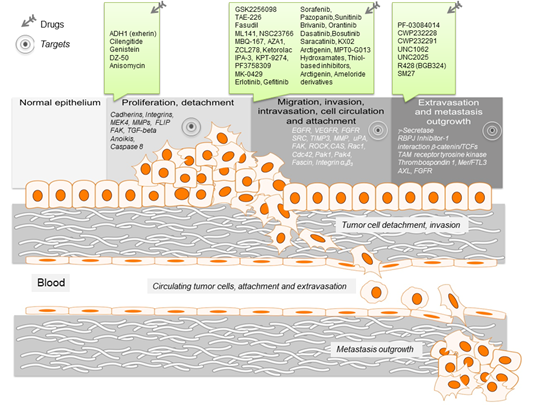
Figure 1. Schematic representation of the key events leading to metastasis. For the different metastatic steps important genes are listed, which represent drivers of the metastatic process. They enable cancer cell migration, invasion intra- and extravasation as well as metastasis outgrowth. Such genes represent promising targets for therapeutic interventions. In this regard, small molecule inhibitors are listed, which target particular steps in the metastatic process.
2. Targets for Therapeutic Intervention during Metastasis Formation
2.1. Tumor Cell Detachment—Principiis Obsta
Cell detachment from the primary tumor has been considered as initial step of metastasis . The adhesion between cells but also to the ECM, is mediated through cell adhesion molecules (CAMs) [11]. Among them, calcium-dependent receptors such as cadherins and integrins play crucial roles. Their dysregulation causes the impairment of tissue integrity [11][12][13].
Cadherins are one group of the CAMs, which are crucial for proper cell-cell contact. Their dysregulation leads to loose cell-cell contacts allowing tumor cells to detach from the primary tumor and disseminate to a distant site [14][15]. For example, the expression of N-cadherin is elevated in many cancer cells and aberrant regulation of this molecule allows them to migrate and form metastases [16][17].
Integrins are further essential CAMs providing cell-ECM interactions [18]. While the extracellular domain of integrins binds to ECM molecules, the intracellular domain facilitates the attachment to the cytoskeleton via intracellular focal adhesions [19]. This binding not only regulates the cell adhesion but also provides the signal transduction between the cell and ECM via integrin activated signal molecules such as focal adhesion kinases (FAKs) and phosphatidylinositol 3-kinase (PI3K) [20]. The aberrant regulation of integrin increases cancer invasiveness via the dysregulation of these signal molecules. It also leads to the activation of matrix metalloproteinases (MMPs) responsible for ECM degradation [21][22]. MMPs are endopeptidases playing an essential role in physiological processes such as organogenesis, apoptosis and cell proliferation [86,87]. Their aberrant regulation leads to tissue damages, enables cancer cell motility and correspondingly causes spread of cell from primary tumors to distant sites [23][24][25].
Physiologically, when cells lost their cell-cell and ECM connection, an apoptotic process called anoikis is activated [26][27]. This process prevents survival and anchorage-independent growth of detached cells and thus hinders dissemination to distant sites. However, some cancer cells develop resistance mechanisms against this control mechanism.
The resistance to anoikis, together with changes in cell adhesion and cell polarity, is conjointly known as epithelial to mesenchymal transition (EMT). This process induces mesenchymal properties of cancer cells leading to increased motility and invasiveness.
These briefly described processes have prompted efforts to therapeutically target the detachment of cells from the primary tumors. They are focusing on three levels, to intervene in the cell detachment and survival of detached cells—(i) targeting CAMs, (ii) activating anoikis and (iii) breaking anoikis resistance.
CAMs represent one of the prominent targets to prevent metastasis initiation. ADH1 (exherin) inhibits the invasion and proliferation of some cancer types through binding and blocking the essential CAM component N-cadherin. It was tested in phase II clinical trials as monotherapy of different N-cadherin positive neoplasms. In clinical phase I settings the combination with cisplatin and gemcitabine for treatment of metastatic pancreatic or biliary tract cancerwas tested, which in part led to stable disease .
Integrins are another prominent class of targets to prevent distant dissemination of tumor cells. The inhibition of integrin–ligand interaction not only decreases cellular growth but also induces apoptotic cell death. The integrin antagonist cilengitide, a cyclic pentapeptide, binds RGD (Arg-Gly-Asp)-dependent integrins, inhibits the ECM ligand-integrin interaction and thus induces apoptosis. The efficacy of this drug was tested in a clinical phase III trial for glioblastoma. In combination with the alkylating agent temozolomide and chemoradiotherapy the application of cilengitide however did not improve patient outcome . By contrast, in a clinical phase I study cilengitide treatment showed an antitumor activity in combination with paclitaxel . In a clinical phase II trial treating non-small lung cancer patients, cilengitide monotherapy was as effective as docetaxel .
Detachment of cancer cells from the ECM leads to conformational changes of integrin, followed by the transmission of outside-to-inside signals through pathways involving FAK. In particular cancer cells, which have high anoikis resistance show an elevated level of FAK expression [28]. Its inhibition by the isoflavanoid genistein reduced detachment of tumor cells and inhibited MMPs. Genistein is tested in different clinical trials, including phase III [NCT00584532] clinical trial for prostate cancer
Anoikis prevents survival of cells that lost their cell-matrix or cell-cell interactions . Therefore, inducing anoikis by drug therapy is another promising approach to reduce the survival of detached cells. DZ-50 is a quinazoline-based compound that inhibits the epithelial and endothelial cell survival through inhibition of surface integrin β1. It reduces the tumor cell adhesion to the ECM by promoting anoikis and thus inhibits tumor growth in vivo .
One of the main pathways leading to the induction of anoikis is the death receptor pathway. This pathway is activated through binding of FAS Cell Surface Death Receptor (FAS) or TNF-Related Apoptosis-Inducing Ligand (TRAIL) to the extracellular domain of the death receptor. This leads to the activation of caspase 8, which cleaves downstream effectors, such as caspase 3 and 7 to finally induce cell death. Caspase 8 and FLICE inhibitory protein (FLIP) are structurally similar proteins. FLIP binds to the DISC complex and inhibits caspase 8 activation [29]. In malignant cells with metastatic potential, FLIP expression is increased, protecting cells from apoptosis. The antibiotic anisomycin (flagecidin) was identified as FLIP inhibitor in a compound library screen . Thus, targeting FLIP with anisomycin leads to anoikis sensitization. The anoikis activating effect of this small molecule was not only shown in vitro for prostate cancer cells but its anti-metastatic effect was also corroborated in mouse studies[29] . Recently, a novel, first-in-class FLIP inhibitor was identified by molecular modelling. The respective lead compounds entered preclinical validation and characterization[30].
Cell detachment and survival of the detached cells are the initial steps of metastatic dissemination. Therefore, it is crucial to target these pathways to prevent dissemination of cells to distant sites.
2.2. Migration of Tumor Cells—Stop Moving
During malignant progression, tumor cells polarize towards chemoattractant gradients and engage in remodeling of the cytoskeleton to physically move away from the primary tumor [31][32]. This requires directed interaction with the ECM via transmembrane receptors (integrins, discoidin domain receptors) for ECM proteins (fibronectin, fibrinogen, collagen, etc.) [33][34]. Upon contact with ECM these receptors form focal adhesions, which in turn activate FAK with assistance by adapter proteins (talin, paxillin). FAK is an important hub of intracellular signaling and integrates integrin and growth hormone receptor signals to various target proteins. In cell migration, FAK orchestrates PI3K/AKT and Rho-GTPase signaling to exert polarized cell motility [35][36][37].
Rho, Rac and cell division cycle 42 (Cdc42) initiate and catalyze the polymerization of actin filaments during lamellipodia and filopodia formation [38][39]. RhoA recruits formin mDia, while Rac and Cdc42 recruit WASP proteins and the Arp2/3 complex [40][41]. The Rock proteins are effectors for Rho, while Cdc42 acts via myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK) to foster actomyosin contractility for effective locomotion of the migrating cell [42]. Downstream of Rac/Cdc42, the family of p21-activated kinases (PAK) increases focal adhesion turnover and LIMK1-dependent actin depolymerization, resulting in cytoskeleton remodeling and migration [43][44].
Another druggable target involved in actomyosin remodeling, Fascin, ties up actin filaments during filopodia formation and crucially contributes to tumor cell motility in vitro and in vivo [45][46][47][48]. Fascin is a negative prognostic marker of cancer patient survival [49][50]. Certain members of small calcium-binding S100 proteins, most notably S100A4, have emerged as accomplice of cancer progression. S100A4 induces cell migration as a catalyst of interaction with F-actin and of myosin-IIa disassembly [51][52][53][54].
Due to its prominent position in cancer progression, FAK has been experimentally targeted with various inhibitors [55]. GSK2256098 intercepts the phosphorylation of FAK at tyrosine residue Y397 and delays pancreatic cancer cell wound closure in vitro . This compound was recently tested clinically in glioblastoma [NCT01138033] and mesothelioma. It prolonged survival of patients with merlin-negative tumors [NCT01938443] [56].
The ATP-competitive FAK inhibitor TAE-226 also exerts efficacy against IGFR1 and was found to restrict glioma cell viability and motility in vitro . Only recently, TAE-226 was able to prevent lung metastasis of orthotopically injected murine breast cancer cells in syngeneic mice [57].
Fasudil (HA1077) directly inhibits ROCK as an effector of RhoA and is already clinically approved in Japan for vasospasms due to its capability to suppress actin stress fiber formation and vascular muscle cell migration . This finding was successfully recapitulated in cancer cells [58][59][60]. However, no clinical trial on cancer has considered fasudil so far.
Multiple novel Rho-GTPase inhibitors prevent pro-migratory cytoskeletal rearrangement and cell motility [61]. While RhoA inhibitors have not been promoted beyond biochemical analyses, Rac1 and Cdc42 inhibitors were promising in restricting cell migration in vitro [62]. Several molecules have been designed to compete with the nucleotide-binding pocket of Rac1 and Cdc42. EHT-1864 is particularly effective in preventing estrogen- and androgen-dependent Rac activation in breast and prostate cancer but might only serve as a prototype due to its adverse effect of platelet apoptosis in mice [63][64][65]. CID2950007 (ML141) and CID44216842 specifically target Cdc42 and restrict the motility of ovarian cancer cells .
Other compounds interfere with guanine nucleotide exchange factor (GEF) binding to Rac/Cdc42 directly. NSC23766 was designed to occupy Rac1’s binding pocket for the GEFs Trio and Tiam [26] and inhibited lamellipodia formation in lung cancer cells [66]. Its unacceptable toxicity, however, stimulated several optimizations leading to EHop-016, MBQ-167 and AZA1 [67].
Therefore, PAK1 as a major executor of Rac1-mediated migration, was targeted in an extensive in silico screen. Two compounds structurally unrelated to NSC23766 emerged as potent targeting pancreatic cancer cell migration while non-toxic towards normal pancreatic cells [68].
A variety of Cdc42 inhibitors have been established. ZCL278 intercepts activation of Cdc42 by Intersectin. It was effective in blocking actin-dependent migration of prostate cancer cells in vitro while having no effect on cell viability . AZA197, derived from AZA1, occupies the nucleotide binding pocket of Cdc42 and prevented colon cancer cell motility and xenograft implantation in vivo.
Despite promising effects in preclinical studies, none of the small molecules discussed have successfully advanced to testing in humans [124,130]. Nevertheless, the R-enantiomer of the common NSAID ketorolac was repurposed in ovarian cancer cells to decrease Cdc42 dependent filopodia formation and cell migration. In a clinical phase III trial the drug was tested for high risk breast cancer patient treatment.
Extensive effort has also been put in the deployment of PAK inhibitors, particularly targeting PAK1 and PAK4 [69]. IPA-3, the only isoform-selective, allosteric inhibitor of PAK1, stabilizes PAK1 in its autoinhibitory state. Thereby, PAK1-dependent cell membrane ruffling is blocked [70]. IPA-3 prevents PAK1 dependent recruitment of WAVE2 and lamellipodia formation. This results in reduced migration and metastasis of i.v.-injected esophageal cancer cells [71][72]. KPT-9274 and KPT-8752 can reduce PAK4 expression, resulting in inhibition of growth and migration of renal cancer cells . A clinical study gauging the safety of KPT-9274 is currently recruiting (NCT02702492). Another PAK4-inhibitor, PF3758309, was shown to restrict lung cancer cell motility. It was prematurely terminated in a clinical trial due to intolerable adverse effects. This indicates the need for further optimization of this compound (NCT00932126).
Treatments with fascin inhibitors NP-G2-029 and NP-G2-044, which prevent interaction with actin filaments, resulted in reduced migration, invasion and metastasis of breast cancer cells [73][74]. NP-G2-044 is currently in a phase I trial in patients with metastatic disease [NCT03199586].
Integrins as mediators of cell migration also support tumor cells in circulation (CTCs) and metastatic settlement in distant organs. Integrin αvβ3 is instrumental for the extravasation of breast cancer cells. Tumor cells recruit platelets within the capillary system of the metastatic site, which in turn release a wide variety of tissue-remodeling factors and facilitate trans-endothelial migration[75]. MK-0429, an orally bioavailable small molecule, prevents lung metastasis of i.v. injected melanoma cells and is a major contestant against mainly antibody-based integrin inhibition [76].
Despite improvements in surgical techniques, that seem to obviate any use of migration inhibition of primary tumors, the consideration of the molecules listed above could have merit in the treatment of unresectable malignancies or in for example, neo-adjuvant settings.
2.3. Invasion Intervention—Stop the Invaders
Invasion through ECM, intravasation to vasculature and extravasation at the distant site of tumor cells is regulated by complex signaling. It involves formation of invadopodia, secretion of proteases as well as factors that attract tumor cells to the metastatic site. In this context, factors that constitute the metastatic niche and the proper environment are essential to promote tumor cell invasion [77].
An important event in tumor cell invasion is the formation of F-actin-rich invadopodia as membrane protrusions. This ensures cellular movement and invasion through the ECM [78]. Invadopodia are important to clear the tumor cell path by degradation of cell-cell junctions and of the ECM. Formation of invadopodia is triggered by growth factors such as EGF, PDGF, basic FGF and also by integrins. These extracellular stimuli activate PI3K and Src signaling leading to actin re-modeling. This is essential to provide the mechanical forces of cell movements [140,141]. Particularly Src signaling has been identified as key event in this process. This is further supported by adhesion domains, which bind to the ECM and provide the anchor promoting directed movement. Further, activity of invadopodia is associated with the action of MMPs and of serine proteases for effective invasion [143,144]. Serine proteases such as uPA not only degrade the ECM but are also known to proteolytically activate growth factors, for example, HGF, TGF-alpha or basic FGF .
These briefly introduced processes of tumor cell invasion are targets for therapeutic intervention. Considering the sequence of events impacting tumor cell invasion, three levels are useful for invasion intervention, which also affect invadopodia formation—(i) stimulation by growth factors, (ii) invasion-promoting signaling and (iii) protease activation.
Growth factors play a decisive role in inducing invasive properties of tumor cells [78][79][80][81][82][83][84][85]. Growth factor receptors, such as EGFR, PDGFR, basic FGFR and VEGFR are in focus. Apart from antibody-based interventions, small molecule inhibitors are in clinical use or under development. For EGFR-signaling erlotinib and gefitinib are known tyrosine kinase inhibitors, whereas sorafenib, sunitinib and pazopanib are inhibitors of the VEGF receptor [38–41]. Further, FGFR and also the VEGFR function is antagonized by brivanib (BMS-582664), a prodrug, which is converted to the ATP-competitor BMS-450215. PDGFR autophosphorylation and therefore receptor activation can be inhibited by the adenine mimetic drug orantinib (SU6668), which also acts on FGF-1, due to structural similarities of the ATP binding sites of the two receptors as target motive for the drug . For all these, mostly multitarget inhibitors, anti-metastatic and anti-invasion activity has been demonstrated.
As mentioned, Src and Src-signaling play an important role in invasion. Its inhibition was shown to intervene in invasion and metastasis. Small molecule inhibitors, which interfere with Src activity and signaling, are the clinically used drugs dasatinib and bosutinib (SKI-606), the dual kinase inhibitor saracatinib (AZD-0530) and the dual Src/tubulin inhibitors KX02, KX2-391. Among those, dasatinib has been shown to inhibit tumor growth and metastasis formation of an orthotopic prostate cancer model . This was associated with drug-mediated reduction in cancer cell migration and invasion. Saracatinib inhibits the invadopodia regulatory proteins FAK, p130 Crk-associated substrate (CAS) and contactin in HNSCC. Such small molecule inhibitors of pathways, which are essential for invadopodia formation and function, indicate the effectiveness of intervention strategies at this driving step of cancer metastasis.
The main function of invadopodia for cancer cells is promotion of matrix degradation to support cancer cell invasion. In this regard targeting proteases is an additional level for effective intervention and prevention of metastasis. In this context, MMPs (e.g., MMP-2, MMP-9) are valuable targets. MMPs are members of zinc-endopeptidases with proteolytic activity against a broad spectrum of ECM substrates to support tumor cell invasion [86][87]. Tumor cells express MMPs at their leading edges to degrade collagen fibers to open the invasion path. Based on this, inhibition of invasion by MMP interference might contribute to inhibition of the entire metastatic process. For such an approach, numerous small molecule inhibitors have been developed. They belong to the group of hydroxamates (batimastat, marimastat, prinomastat, solimastat etc.), thiol-based MMP inhibitors (e.g., rebimastat, tanomastat) and other MMP inhibitors, such as carbamoylphosphonate cis-ACCP, pyrimidine-trione-like Ro28-2653 or the sulfonamide derivative S-3304. Clinical testing of such inhibitors however revealed low efficacy of MMP intervention. This is potentially due to low selectivity of the drugs or due to emergence of resistance mechanisms in treated tumors. Another resistance mechanism is the switch of tumor cells from protease-dependent to protease-independent invasion to circumvent the inhibitory activity of applied small molecule drugs. This is the rational, by which the tissue inhibitors of metalloproteases (TIMPs) came into focus. Here, small molecules are of increasing interest, which stimulate TIMP expression to inhibit the malicious action of MMPs in tumor invasion and metastasis. Such activity has been tested for compounds like the organo-sulfur compound diallyl-disulfide, the lignan arctigenin or the arylsulfonamide derivative MPT0G013 and many others, which all increased TIMP-3 expression and inhibited tumor cell migration and invasion.
Due to the migration, invasion and metastasis promoting function of the serine protease uPA, numerous approaches are aiming at inhibition of its proteolytic activity [88]. Small molecule inhibitors, such as the ameloride derivatives B-428 and B-623 were shown to inhibit prostate and also breast cancer growth and metastasis in vivo . Similarly, encouraging results have been obtained by testing WX-671 and its prodrug WX-UK1 in clinical trials for treatment of solid tumors . Such uPA targeting approaches indicate the therapeutic value for tumor suppression and metastasis reduction.
In summary, there is a plethora of small molecules available, which act on processes of invadopodia action, cell migration and invasion. Their use for intervention in signaling processes of migration and invasion and combination with other drugs that interfere in other essential steps of metastasis formation will contribute to improved anti-metastatic therapy of cancer.
References
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757, doi:10.1016/S0140-6736(07)60781-8.
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695, doi:10.1016/j.cell.2006.11.001.
- Chaffer, C.L.; A Weinberg, R. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564, doi:10.1126/science.1203543.
- Budczies, J.; Kluck, K.; Walther, W.; Stein, U. Decoding and targeting the molecular basis of MACC1-driven metastatic spread: Lessons from big data mining and clinical-experimental approaches. Semin. Cancer Boil. 2020, 60, 365–379, doi:10.1016/j.semcancer.2019.08.010.
- Zijlstra, A.; The Board Members of the Metastasis Research Society; Von Lersner, A.; Yu, D.; Borrello, L.; Oudin, M.; Kang, Y.; Sahai, E.; Fingleton, B.; Stein, U. The importance of developing therapies targeting the biological spectrum of metastatic disease. Clin. Exp. Metastasis 2019, 36, 305–309, doi:10.1007/s10585-019-09972-3.
- Valastyan, S.; A Weinberg, R. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292, doi:10.1016/j.cell.2011.09.024.
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456, doi:10.1038/nrc1370.
- Fidler, I.J.; Poste, G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008, 9, 808, doi:10.1016/s1470-2045(08)70201-8.
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109, doi:10.1080/14737140.2020.1718496.
- Anderson, R.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204, doi:10.1038/s41571-018-0134-8.
- Li, D.-M.; Feng, Y.-M. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: Potential therapeutic targets. Breast Cancer Res. Treat. 2011, 128, 7–21, doi:10.1007/s10549-011-1499-x.
- Albelda, S.M. Role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab. Investig. 1993, 68, 4–17.
- Aplin, A.E.; Howe, A.; Alahari, S.K.; Juliano, R.L. Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol. Rev. 1998, 50, 197–263.
- Cavallaro, U.; Christofori, G. Cell adhesion in tumor invasion and metastasis: Loss of the glue is not enough. Biochim. Biophys. Acta (BBA) Bioenerg. 2001, 1552, 39–45, doi:10.1016/s0304-419x(01)00038-5.
- Alizadeh, A.M.; Shiri, S.; Farsinejad, S. Metastasis review: From bench to bedside. Tumor Boil. 2014, 35, 8483–8523, doi:10.1007/s13277-014-2421-z.
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous Expression of N-Cadherin in Breast Cancer Cells Induces Cell Migration, Invasion, and Metastasis. J. Cell Boil. 2000, 148, 779–790, doi:10.1083/jcb.148.4.779.
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-Cadherin Promotes Motility in Human Breast Cancer Cells Regardless of Their E-Cadherin Expression. J. Cell Boil. 1999, 147, 631–644, doi:10.1083/jcb.147.3.631.
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22, doi:10.1038/nrc2748.
- Hamidi, H.; Pietilä, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023, doi:10.1038/bjc.2016.312.
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Mol. Asp. Med. 2010, 31, 205–214, doi:10.1016/j.mam.2010.02.001.
- I Deryugina, E.; A Bourdon, M.; Luo, G.X.; A Reisfeld, R.; Strongin, A. Matrix metalloproteinase-2 activation modulates glioma cell migration. J. Cell Sci. 1997, 110, 110.
- Brooks, P.C.; Stromblad, S.; Sanders, L.C.; Von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; A Cheresh, D. Localization of Matrix Metalloproteinase MMP-2 to the Surface of Invasive Cells by Interaction with Integrin αvβ3. Cell 1996, 85, 683–693, doi:10.1016/s0092-8674(00)81235-0.
- Charles, J.M. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front. Biosci. 2006, 11, 1696, doi:10.2741/1915.
- John, A.; Tuszynski, G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol. Oncol. Res. 2001, 7, 14–23, doi:10.1007/bf03032599.
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Boil. 2015, 44, 200–206, doi:10.1016/j.matbio.2015.01.019.
- Frisch, S.M.; Screaton, R. Anoikis mechanisms. Curr. Opin. Cell Boil. 2001, 13, 555–562, doi:10.1016/s0955-0674(00)00251-9.
- Gilmore, A.P. Anoikis. Cell Death Differ 2005, 12 (Suppl. 2), 1473–1477, doi:10.1038/sj.cdd.4401723.
- Duxbury, M.; Ito, H.; Zinner, M.; Ashley, S.; Whang, E. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery 2004, 135, 555–562, doi:10.1016/j.surg.2003.10.017.
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Boil. 2012, 2012, 1–11, doi:10.1155/2012/306879.
- Higgins, C.A.; Fox, J.; Majkut, J.; Fiedler, G.E.; Roberts, J.; Humphreys, L.; Boffey, R.J.; Perrior, T.R.; Harrison, T.; Longley, D.B. Abstract 382: Development and pre-clinical assessment of a first-in-class small molecule inhibitor of FLIP. Exp. Mol. Ther. 2019, 79, 382–382, doi:10.1158/1538-7445.am2019-382.
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587, doi:10.1038/nrc3078.
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261, doi:10.4161/cam.23840.
- Rammal, H.; Saby, C.; Magnien, K.; Van Gulick, L.; Garnotel, R.; Buache, E.; El Btaouri, H.; Jeannesson, P.; Morjani, H. Discoidin Domain Receptors: Potential Actors and Targets in Cancer. Front. Pharmacol. 2016, 7, 1321, doi:10.3389/fphar.2016.00055.
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691.
- McLean, G.W.; Carragher, N.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515, doi:10.1038/nrc1647.
- Serrels, B.; Sandilands, E.; Serrels, A.; Baillie, G.; Houslay, M.; Brunton, V.G.; Canel, M.; Machesky, L.M.; Anderson, K.; Frame, M. A Complex between FAK, RACK1, and PDE4D5 Controls Spreading Initiation and Cancer Cell Polarity. Curr. Boil. 2010, 20, 1086–1092, doi:10.1016/j.cub.2010.04.042.
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Boil. 2004, 265, 23–32, doi:10.1016/j.ydbio.2003.06.003.
- Durand-Onaylı, V.; Haslauer, T.; Härzschel, A.; Hartmann, T.N. Rac GTPases in Hematological Malignancies. Int. J. Mol. Sci. 2018, 19, 4041, doi:10.3390/ijms19124041.
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76, doi:10.1007/s10555-008-9170-7.
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12, doi:10.1038/nrm3492.
- Wilkinson, S.; Paterson, H.F.; Marshall, C.J. Cdc42–MRCK and Rho–ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nature 2005, 7, 255–261, doi:10.1038/ncb1230.
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25, doi:10.1038/nrc3645.
- King, H.; Nicholas, N.S.; Wells, C.M. Role of p-21-Activated Kinases in Cancer Progression. Int. Rev. Cell Mol. Biol. 2014, 309, 347–387, doi:10.1016/b978-0-12-800255-1.00007-7.
- Schoumacher, M.; El-Marjou, F.; Laé, M.; Kambou, N.; Louvard, D.; Robine, S.; Vignjevic, D. Conditional expression of fascin increases tumor progression in a mouse model of intestinal cancer. Eur. J. Cell Boil. 2014, 93, 388–395, doi:10.1016/j.ejcb.2014.08.002.
- Vignjevic, D.; Schoumacher, M.; Gavert, N.; Janssen, K.-P.; Jih, G.; Laé, M.; Louvard, D.; Ben-Ze’Ev, A.; Robine, S. Fascin, a Novel Target of β-Catenin-TCF Signaling, Is Expressed at the Invasive Front of Human Colon Cancer. Cancer Res. 2007, 67, 6844–6853, doi:10.1158/0008-5472.can-07-0929.
- Claessens, M.M.A.E.; Bathe, M.; Frey, E.; Bausch, A.R. Actin-binding proteins sensitively mediate F-actin bundle stiffness. Nat. Mater. 2006, 5, 748–753, doi:10.1038/nmat1718.
- Jawhari, A.U.; Buda, A.; Jenkins, M.; Shehzad, K.; Sarraf, C.; Noda, M.; Farthing, M.J.G.; Pignatelli, M.; Adams, J.C. Fascin, an Actin-Bundling Protein, Modulates Colonic Epithelial Cell Invasiveness and Differentiation in Vitro. Am. J. Pathol. 2003, 162, 69–80, doi:10.1016/s0002-9440(10)63799-6.
- Li, A.; Morton, J.P.; Ma, Y.; Karim, S.A.; Zhou, Y.; Faller, W.; Woodham, E.F.; Morris, H.T.; Stevenson, R.P.; Juin, A.; et al. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology 2014, 146, 1386–1396.e1, doi:10.1053/j.gastro.2014.01.046.
- Tan, V.; Lewis, S.; Adams, J.C.; Martin, R.M. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: A systematic review and meta-analysis. BMC Med. 2013, 11, 52, doi:10.1186/1741-7015-11-52.
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629, doi:10.1007/s12551-018-0471-y.
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109, doi:10.1038/nrc3893.
- Stein, U.; Arlt, F.; Walther, W.; Smith, J.; Waldman, T.; Harris, E.D.; Mertins, S.D.; Heizmann, C.W.; Allard, D.; Birchmeier, W.; et al. The Metastasis-Associated Gene S100A4 Is a Novel Target of β-catenin/T-cell Factor Signaling in Colon Cancer. Gastroenterology 2006, 131, 1486–1500, doi:10.1053/j.gastro.2006.08.041.
- Takenaga, K.; Nakamura, Y.; Sakiyama, S.; Hasegawa, Y.; Sato, K.; Endo, H. Binding of pEL98 protein, an S100-related calcium-binding protein, to nonmuscle tropomyosin. J. Cell Boil. 1994, 124, 757–768, doi:10.1083/jcb.124.5.757.
- Roy-Luzarraga, M.; Hodivala-Dilke, K. Molecular Pathways: Endothelial Cell FAK-A Target for Cancer Treatment. Clin. Cancer Res. 2016, 22, 3718–3724, doi:10.1158/1078-0432.CCR-14-2021.
- Mak, G.; Soria, J.-C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981, doi:10.1038/s41416-019-0452-3.
- Fukami, S.; Tomioka, D.; Murakami, Y.; Honda, T.; Hatakeyama, S. Pharmacological profiling of a dual FAK/IGF-1R kinase inhibitor TAE226 in cellular and in vivo tumor models. BMC Res. Notes 2019, 12, 347, doi:10.1186/s13104-019-4389-7.
- Carboni, S.S.C.M.; Lima, N.A.R.; Pinheiro, N.M.; Murta, B.M.T.; Crema, V.O. HA-1077 inhibits cell migration/invasion of oral squamous cell carcinoma. Anti Cancer Drugs 2015, 26, 1–930, doi:10.1097/cad.0000000000000267.
- Yang, X.; Liu, Y.; Zong, Z.; Tian, D. The Rho kinase inhibitor fasudil inhibits the migratory behaviour of 95-D lung carcinoma cells. Biomed. Pharmacother. 2010, 64, 58–62, doi:10.1016/j.biopha.2009.08.006.
- Ying, H.; Biroc, S.L.; Li, W.-W.; Alicke, B.; Xuan, J.-A.; Pagila, R.; Ohashi, Y.; Okada, T.; Kamata, Y.; Dinter, H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006, 5, 2158–2164, doi:10.1158/1535-7163.mct-05-0440.
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111, doi:10.1158/0008-5472.can-18-0619.
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010, doi:10.1517/17460441.2015.1058775.
- Dutting, S.; Heidenreich, J.; Cherpokova, D.; Amin, E.; Zhang, S.-C.; Ahmadian, M.R.; Brakebusch, C.; Nieswandt, B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015, 13, 827–838, doi:10.1111/jth.12861.
- Castoria, G.; D’Amato, L.; Ciociola, A.; Giovannelli, P.; Giraldi, T.; Sepe, L.; Paolella, G.; Barone, M.V.; Migliaccio, A.; Auricchio, F. Androgen-Induced Cell Migration: Role of Androgen Receptor/Filamin A Association. PLoS ONE 2011, 6, e17218, doi:10.1371/journal.pone.0017218.
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and Mechanism of Action of EHT 1864, a Novel Small Molecule Inhibitor of Rac Family Small GTPases. J. Boil. Chem. 2007, 282, 35666–35678, doi:10.1074/jbc.m703571200.
- Xu, L.-Q.; Chen, Q.-Y.; Jiao, D.-M.; Yao, Q.-H.; Wang, Y.-Y.; Hu, H.-Z.; Wu, Y.-Q.; Song, J.; Yan, J. Silencing of Rac1 modifies lung cancer cell migration, invasion and actin cytoskeleton rearrangements and enhances chemosensitivity to antitumor drugs. Int. J. Mol. Med. 2011, 28, 769–776, doi:10.3892/ijmm.2011.775.
- Maes, H.; Van Eygen, S.; Krysko, D.V.; Vandenabeele, P.; Nys, K.; Rillaerts, K.; Garg, A.D.; Verfaillie, T.; Agostinis, P. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014, 5, e1127, doi:10.1038/cddis.2014.94.
- Arnst, J.L.; Hein, A.L.; Taylor, M.A.; Palermo, N.Y.; Contreras, J.I.; Sonawane, Y.A.; Wahl, A.O.; Ouellette, M.M.; Natarajan, A.; Yan, Y. Discovery and characterization of small molecule Rac1 inhibitors. Oncotarget 2017, 8, 34586–34600, doi:10.18632/oncotarget.16656.
- Rane, C.K.; Minden, A. P21 activated kinase signaling in cancer. Semin. Cancer Boil. 2019, 54, 40–49, doi:10.1016/j.semcancer.2018.01.006.
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.E.; Myers, C.; Chernoff, J.; Peterson, J. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Boil. 2008, 15, 322–331, doi:10.1016/j.chembiol.2008.03.005.
- Chen, L.; Bi, S.; Hou, J.; Zhao, Z.; Wang, C.; Xie, S. Targeting p21-activated kinase 1 inhibits growth and metastasis via Raf1/MEK1/ERK signaling in esophageal squamous cell carcinoma cells. Cell Commun. Signal. 2019, 17, 31, doi:10.1186/s12964-019-0343-5.
- Takahashi, K.; Suzuki, K. Membrane transport of WAVE2 and lamellipodia formation require Pak1 that mediates phosphorylation and recruitment of stathmin/Op18 to Pak1–WAVE2–kinesin complex. Cell. Signal. 2009, 21, 695–703, doi:10.1016/j.cellsig.2009.01.007.
- Han, S.; Huang, J.; Liu, B.; Xing, B.; Bordeleau, F.; Reinhart-King, C.A.; Li, W.; Zhang, J.J.; Huang, X.-Y. Improving fascin inhibitors to block tumor cell migration and metastasis. Mol. Oncol. 2016, 10, 966–980, doi:10.1016/j.molonc.2016.03.006.
- Huang, F.-K.; Han, S.; Xing, B.; Huang, J.; Liu, B.; Bordeleau, F.; Reinhart-King, C.A.; Zhang, J.J.; Huang, X.-Y. Targeted inhibition of fascin function blocks tumour invasion and metastatic colonization. Nat. Commun. 2015, 6, 7465, doi:10.1038/ncomms8465.
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134, doi:10.1038/nrc3004.
- Raab-Westphal, S.; Marshall, J.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110, doi:10.3390/cancers9090110.
- Goubran, H.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of Tumor Growth and Metastasis: The Role of Tumor Microenvironment. Cancer Growth Metastasis 2014, 7, CGM.S11285–18, doi:10.4137/cgm.s11285.
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Boil. 2015, 3, 4, doi:10.3389/fcell.2015.00004.
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2013, 33, 4193–4202, doi:10.1038/onc.2013.393.
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J.S. Spatial regulation of tumor cell protrusions by RhoC. Cell Adhes. Migr. 2014, 8, 263–267, doi:10.4161/cam.28405.
- Madsen, M.A.; Deryugina, E.I.; Niessen, S.; Cravatt, B.F.; Quigley, J.P. Activity-based Protein Profiling Implicates Urokinase Activation as a Key Step in Human Fibrosarcoma Intravasation. J. Boil. Chem. 2006, 281, 15997–16005, doi:10.1074/jbc.m601223200.
- Moss, L.A.S.; Jensen-Taubman, S.; Stetler-Stevenson, W.G. Matrix Metalloproteinases. Am. J. Pathol. 2012, 181, 1895–1899, doi:10.1016/j.ajpath.2012.08.044.
- Xue, C.; Wyckoff, J.; Liang, F.; Sidani, M.; Violini, S.; Tsai, K.-L.; Zhang, Z.Y.; Sahai, E.; Condeelis, J.S.; E Segall, J. Epidermal Growth Factor Receptor Overexpression Results in Increased Tumor Cell MotilityIn vivoCoordinately with Enhanced Intravasation and Metastasis. Cancer Res. 2006, 66, 192–197, doi:10.1158/0008-5472.can-05-1242.
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325, doi:10.1023/A:1023768811842.
- Kedrin, D.; Wyckoff, J.; Boimel, P.; Coniglio, S.J.; Hynes, N.E.; Arteaga, C.L.; E Segall, J. ERBB1 and ERBB2 have distinct functions in tumor cell invasion and intravasation. Clin. Cancer Res. 2009, 15, 3733–3739, doi:10.1158/1078-0432.CCR-08-2163.
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573, doi:10.1016/j.cardiores.2005.12.002.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370, doi:10.3389/fonc.2019.01370.
- McMahon, B.J.; Kwaan, H.C. Components of the Plasminogen-Plasmin System as Biologic Markers for Cancer. Adv. Exp. Med. Biol. 2015, 867, 145–156, doi:10.1007/978-94-017-7215-0_10.
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nature 2013, 15, 807–817, doi:10.1038/ncb2767.

