 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Raquel Sanabria de la Torre | -- | 2651 | 2022-06-30 07:56:10 | | | |
| 2 | Dean Liu | + 35 word(s) | 2686 | 2022-07-04 07:43:08 | | |
Video Upload Options
Vascular complications are the leading cause of morbidity and mortality among patients with type 2 diabetes mellitus (T2DM). These vascular abnormalities result in a chronic hyperglycemic state, which influences many signaling molecular pathways that initially lead to increased oxidative stress, increased inflammation, and endothelial dysfunction, leading to both microvascular and macrovascular complications. Endothelial dysfunction represents the initial stage in both types of vascular complications; it represents “mandatory damage” in the development of microvascular complications and only “introductory damage” in the development of macrovascular complications. Increasing scientific evidence has revealed an important role of the Wnt pathway in the pathophysiology of the vascular wall. It is well known that the Wnt pathway is altered in patients with T2DM.
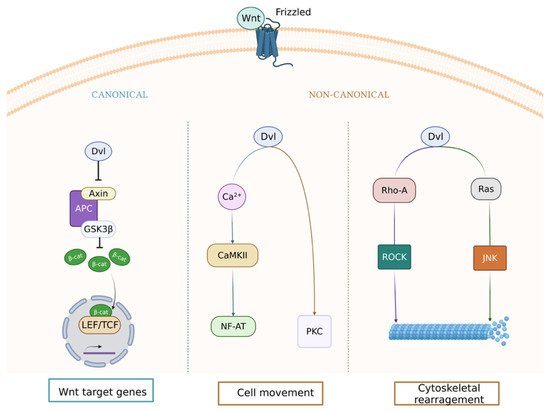
1. Wnt Signaling Pathway in the Vasculature
2. Wnt Pathway and Microvascular Disease in Type 2 Diabetes Mellitus
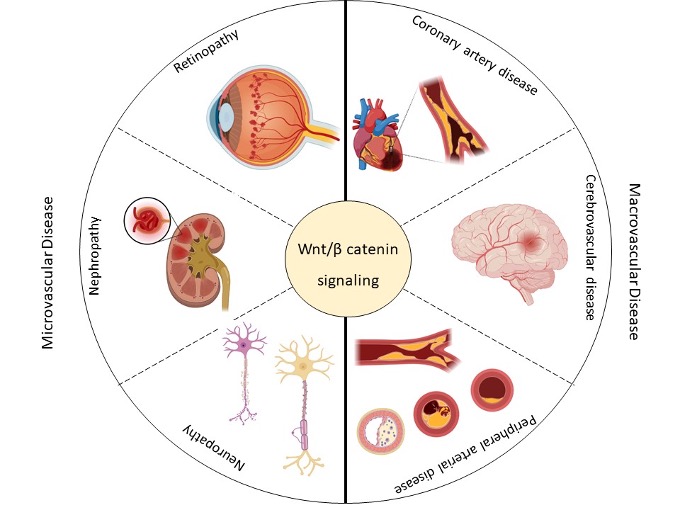
Figure 2. This scheme summarizes the micro- (retinopathy, nephropathy, and neuropathy) and macrovascular (coronary artery disease, cerebrovascular disease, and peripheral arterial disease) complications associated with type 2 diabetes mellitus (T2DM) and their possible association with the Wingless-Int (Wnt) signaling pathway. Created with BioRender.com.
3. Wnt Pathway and Microvascular Disease in Type 2 Diabetes Mellitus
Table 1. Components of the Wnt pathway that can be altered in the microvascular complications of T2DM.1
| Disease | Event | Component | Expression | In Vitro | In Vivo | Reference |
|---|---|---|---|---|---|---|
| Microvascular | Retinopathy | β-catenin | ↑ | Inflammation and angiogenesis | Retinal inflammation and vascular leakage | [20] |
| LRP5/6 | ↑ | Inflammation and angiogenesis | Retinal inflammation and vascular leakage | [20] | ||
| ↓ | Lack of deeper retinal vessels | Significant decrease in pathological retinal neovascularization Significant decrease in retinal vascularization during development Affects blood–retinal barrier formation |
[21] | |||
| Dkk1 | ↑ | Inhibition of the generation of reactive oxygen species (ROS) | Mitigated retinal inflammation and blocked overexpression of proinflammatory factors such as ICAM-1 and COX-2 Reduction in retinal vascular leakage and improvement of ischemia-induced retinal neovascularization |
[20] | ||
| Frizzled4 | ↑ | Angiogenesis | Pathological neovascularization | [21] | ||
| Dvl2 | ↓ | Impaired angiogenesis | Significant decrease in pathological retinal neovascularization | [21] | ||
| Claudin-5 | ↓ | Significant suppression of endothelial cell sprouting | Suppression of pathological vascular growth and development | [21] | ||
| Frizzled7 | ↑ | Inflammation, angiogenesis, and oxidative stress | Pathological neovascularization | [22] | ||
| SERPINA3K | ↑ | Inhibition of connective tissue growth factor overexpression | Antioxidation Anti-inflammatory Antifibrosis |
[23] | ||
| VLDLR | ↑ | Anti-angiogenesis Inhibited endothelial cell proliferation, migration, and tube formation |
Improvement of ocular neovascularization, | [24] | ||
| Endostatin | ↑ | Impaired angiogenesis | Reduced VEGF-induced retinal vascular permeability, neovascularization, and retinal detachment | [25] | ||
| Kallistatin | ↑ | Anti-inflammation Anti-angiogenesis |
Attenuation of ischemia-induced retinal neovascularization | [26] | ||
| PEDF | ↑ | Anti-inflammation Anti-angiogenesis |
Ameliorated retinal inflammation, vascular leakage, and neovascularization | [27] | ||
| MiARN-184 | ↑ | Anti-angiogenesis | Improves inflammatory responses, vascular leakage, and neovascularization. | [28] | ||
| Nephropathy | β-catenin | ↑ | Reduced mesangial cell apoptosis Podocyte dysfunction |
Glomerular albuminuria and subsequent glomerular injury | [29] | |
| ↓ | Mesangial cells apoptosis | Increased severity of streptozotocin-induced diabetes nephritis | [29] | |||
| LEF1 | ↑ | Enhanced proliferation and metastasis of renal cells | Renal cell carcinoma (RCC) | [30] | ||
| LRP6 | ↓ | Mesangial cell apoptosis | Attenuated renal inflammation, reduced proteinuria, and ameliorated fibrosis | [31] | ||
| Wnt4 | ↑ | Stimulation of mesenchymal-to-epithelial differentiation Podocyte dysfunction |
Tubulo-interstitial fibrosis Glomerular albuminuria and subsequent glomerular injury |
[29] | ||
| ↓ | Mesangial cell apoptosis | Kidney tissue disorganization, as well as disease development and progression | [32] | |||
| Dkk1 | ↑ | Amelioration of podocyte apoptosis and viability | Restored podocyte function and decreased albuminuriaBone-mineral disorder syndrome | [29][33] | ||
| TRPC6 | ↑ | Podocyte injury | Excessive calcium influx in podocytes leading to foot process effacement, podocyte apoptosis, and subsequent glomerular damage | [29] | ||
| Wnt9a | ↑ | Evoking of cell communication between senescent tubular cells and interstitial fibroblasts | Tubular senescence and renal fibrosis | [34] | ||
| Wnt5a | ↑ | Increased ROS production | Mesangial cell apoptosis | [35] | ||
| CTGF/CCN2 | ↑ | LRP6 phosphorylation and accumulation of β-catenin | Attenuated renal inflammation, reduced proteinuria, and ameliorated fibrosis Mesangial cell apoptosis |
[31] | ||
| CTNNB1 | ↓ | Improved podocyte motility | Damage to the basement membrane, albuminuria, and increased susceptibility to glomerular injury | [35] | ||
| Wnt6 | ↓ | Damaged tubulo-interstitium | Renal fibrosis | [36] | ||
| Neuropathy | PORCN | ↓ | Slightly reduced expression of Wnt3a Significantly reduced expression of β-catenin, Dvl1, c-myc, GRP78, and MMP2 in the sciatic nerve |
Decreased heat- and cold-induced hyperalgesia Increased motor nerve conduction speed Increased sensory nerve conduction speed Increased nerve blood flow Increased density of intraepidermal nerve fibers |
[37] | |
| Dvl | ↓ | Significantly reduced expression of β-catenin, Dvl1, c-myc, GRP78, and MMP2 in the sciatic nerve | Decreased heat- and cold-induced hyperalgesia Increased motor nerve conduction speed Increased sensory nerve conduction speed Increased nerve blood flow Increased density of intraepidermal nerve fibers |
[37] | ||
| β-catenin | ↓ | Significantly reduced expression of β-catenin, Dvl1, c-myc, GRP78, and MMP2 in the sciatic nerve | Decreased heat- and cold-induced hyperalgesia Increased motor nerve conduction speed Increased sensory nerve conduction speed Increased nerve blood flow Increased density of intraepidermal nerve fibers |
[37] | ||
| Wnt3a | ↑ | Release of brain-derived neurotrophic factor in microglial cells | Allodynia | [38] | ||
| XAV939 | ↑ | - | Effective attenuation of neuropathic pain induction Drastic attenuation of the development of allodynia |
[38] |
4. Wnt Pathway and Macrovascular Disease in Type 2 Diabetes Mellitus
| Disease | Event | Component | Expression | In Vitro | In Vivo | Reference |
|---|---|---|---|---|---|---|
| Macrovascular | Coronary artery disease | Scl | ↑ | Endothelial dysfunction, alteration on proliferation, and migration of vascular smooth muscle cells | Atherosclerotic process, abnormal intima-media thickness, carotid plaques, aortic calcifications, and mortality | [44][45] |
| Dkk-1 | ↑ | Regulates platelet-mediated inflammation and contributes to plaque de-escalation | Ischemic stroke and cardiovascular death | [46] | ||
| ↑ | Endothelial activation and release of inflammatory cytokines Endothelial–mesenchymal transition in aortic endothelial cells |
Onset and progression of atherosclerosis | [47] | |||
| LRP6 | ↓ | LDL uptake was significantly lower in lymphoblastoid cells | Elevated plasma cholesterol and elevated plasma LDL, triglyceride, and fatty liver levels | [48] | ||
| Wnt5a | ↑ | Induction of inflammatory gene expression GM-CSF, IL-1a, IL-3, IL-5, IL-6, IL-7, IL-8, CCL2, CCL8, and COX-2 in human aortic endothelial cells | Elevation of triglyceride levels, vascular insulin resistance, and endothelial dysfunction | [49] | ||
| ↑ | Macrophage activation | Increased recruitment of inflammatory cells and amplified inflammatory response | [50] | |||
| Dkk-3 | ↓ | Increased intima-media thickness of the carotid artery | Delayed reendothelialization and aggravated neointima formation | [51] | ||
| ↑ | Induces differentiation of vascular progenitors and fibroblasts into smooth muscle cells | Larger and more vulnerable atherosclerotic lesions with more macrophages, fewer smooth muscle cells, and less extracellular matrix deposition | [52] | |||
| TCF7L2 | ↓ | Loss of differentiation of vascular smooth muscle cells | Medial aortic hyperplasia | [53] | ||
| Wnt2 | ↑ | Regulates smooth muscle cell migration | Triggers intima-media thickening | [54] | ||
| LRP5 | ↓ | Activation of proinflammatory genes (interferon γ, IL15, IL18, and TNF ligand superfamily 13b). | Larger aortic atherosclerotic lesions | [55] | ||
| Cerebrovascular disease | Scl | ↑ | Arterial calcification | Ischemic stroke caused by atherosclerotic stroke of large arteries or occlusion of small arteries | [56] | |
| Dkk1 | ↑ | Biomarker for the presence of coronary atherosclerotic plaque | Carotid atherosclerosis, stable angina, and myocardial infarction Poor prognosis 1 year after ischemic stroke |
[57] | ||
| miR-150-5p | ↑ | Regulates the Wnt signaling pathway and participates in cell proliferation and apoptosis by downregulating p53 | Inhibition of cell proliferation, colony formation, and tumor growth | [58] | ||
| ↓ | CD133− cells acquire a stem-cell-like phenotype | >Glioma | [59] | |||
| β-catenin | ↑ | Key regulators for cadherin-mediated cell–cell adhesion |
Glioma Higher degree of malignancy of the tumor |
[59] | ||
| Wnt1 | ↓ | Neuronal disappearance and increasing functional deficits | Oxidant stress and cerebral ischemia | [60] | ||
| claudin-1 | ↓ | Neuronal damage | Increased permeability of the blood–brain barrier, petechial hemorrhage in the brain, neuronal injury, and central nervous system inflammation | [61] | ||
| Claudin-3 | ↓ | Neuronal damage | Intracerebral petechial hemorrhages | [62] | ||
| Wnt3a | ↑ | Alleviates neuronal apoptosis at the cellular and subcellular levels | Neuroprotection in traumatic brain injury, and ischemic stroke | [63] | ||
| LRP6 | ↓ | Increased expression of inflammatory genes after middle artery occlusion | Risk of ischemic stroke, larger heart attack, and severe motor deficits | [64] | ||
| Wnt5 | ↑ | Enhanced endothelial activation type 1 inflammatory mediator to promote endothelial activation type 2 | Brain aging Inflamed atheroma plaques |
[65] | ||
| miRNA-148b | ↓ | Attenuates neural stem-cell proliferation and differentiation | Reduces ischemic injury and improves neurological function | [66] | ||
| Peripheral arterial disease | Wnt5a | ↑ | Endothelial dysfunction | Increased risk of peripheral arterial occlusive disease, as well as metabolic and cardiovascular disorders | [67] | |
| Sfrp5 | ↓ | Inhibition of cardiac fibroblast proliferation and migration Inflammation and myocardial injury |
ST-segment elevation myocardial infarction, metabolic syndrome, and increased risk of peripheral arterial occlusive disease | [67] | ||
| CTHRC1 | ↑ | Synovial hyperplasia, contributes to the inflammatory microenvironment, and promotes pannus invasion through increased motility and invasion of synoviocytes | Increased risk of systemic lupus erythematosus, development of rheumatoid arthritis, and severity of the disease | [68] | ||
| ALKBH5 | ↑ | Reduced proliferation and migration and decreased viability in hypoxic cardiac microvascular endothelial cells | Impaired hypoxic tube formation, but not the normoxic cardiac microvascular endothelial cells | [69] |
References
- Reis, M.; Liebner, S. Wnt signaling in the vasculature. Exp. Cell Res. 2013, 319, 1317–1323.
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. 2012, 23, 628–636.
- Freese, J.L.; Pino, D.; Pleasure, S.J. Wnt signaling in development and disease. Neurobiol. Dis. 2010, 38, 148–153.
- Jansson, L.; Kim, G.S.; Cheng, A.G. Making sense of Wnt signaling—Linking hair cell regeneration to development. Front. Cell. Neurosci. 2015, 9, 66.
- Catalano, A.; Bellone, F.; Morabito, N.; Corica, F. Sclerostin and vascular pathophysiology. Int. J. Mol. Sci. 2020, 21, 4779.
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.N.; Komm, B.S.; Javed, A.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140.
- Bundy, K.; Boone, J.; Simpson, C.L. Wnt signaling in vascular calcification. Front. Cardiovasc. Med. 2021, 8, 3–8.
- Komori, T. Regulation of proliferation, differentiation and functions of osteoblasts by runx2. Int. J. Mol. Sci. 2019, 20, 1694.
- Duan, P.; Bonewald, L. The role of the Wnt/β-catenin signaling pathway in formation and maintenance of bone and teeth. Int. J. Biochem. Cell Biol. 2016, 77, 23–29.
- Tanaka, S.; Matsumoto, T. Sclerostin: From bench to bedside. J. Bone Miner. Metab. 2021, 39, 332–340.
- Gay, A.; Towler, D.A.; Wilson, M.E.; Opin, C.; Author, L. Wnt signaling in cardiovascular disease: Opportunities and challenges HHS public access author manuscript. Curr. Opin. Lipidol. 2017, 28, 387–396.
- aFGF Alleviates Diabetic Endothelial Dysfunction by Decreasing Oxidative Stress via Wnt/β-Catenin-Mediated Upregulation of HXK2|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S2213231720310168?token=ACF7439396356846411802F2F5DA6A45B3962C9A3A11A6D4E8AAC1B6FB88D7C48A4219F0EC57BA9A38F5B755B2436306&originRegion=eu-west-1&originCreation=20220617104143 (accessed on 17 May 2022).
- Jia, Q.; Zhu, R.; Tian, Y.; Chen, B.; Li, R.; Li, L.; Wang, L.; Che, Y.; Zhao, D.; Mo, F.; et al. Salvia miltiorrhiza in diabetes: A review of its pharmacology, phytochemistry, and safety. Phytomedicine 2019, 58, 152871.
- Huang, L.; Lin, T.; Shi, M.; Chen, X.; Wu, P. Liraglutide suppresses production of extracellular matrix proteins and ameliorates renal injury of diabetic nephropathy by enhancing Wnt/β-catenin signaling. Am. J. Physiol. Renal Physiol. 2020, 319, F458–F468.
- Smits, M.M.; Tonneijck, L.; Muskiet, M.H.A.; Hoekstra, T.; Kramer, M.H.H.; Diamant, M.; Serné, E.H.; Van Raalte, D.H. GLP-1-based therapies have no microvascular effects in type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2125–2132.
- Strain, W.D.; Paldánius, P.M. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2018, 17, 57.
- Rizzoni, D.; Rosei, E.A. Small artery remodeling in hypertension and diabetes. Curr. Hypertens. Rep. 2006, 8, 90–95.
- Valero, K.; Marante, D.; Torres, M.R.; Ramírez, G.; Cortéz, R.; Carlini, R. Complicaciones microvasculares de la diabetes. Rev. Venez. Endocrinol. Metab. 2012, 10, 111–137.
- Summers, M.E.; Richmond, B.W.; Kropski, J.A.; Majka, S.A.; Bastarache, J.A.; Hatzopoulos, A.K.; Bylund, J.; Ghosh, M.; Petrache, I.; Foronjy, R.F.; et al. Balanced Wnt/Dickkopf-1 signaling by mesenchymal vascular progenitor cells in the microvascular niche maintains distal lung structure and function. Am. J. Physiol. Cell Physiol. 2021, 320, C119.
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.X. Activation of the wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am. J. Pathol. 2009, 175, 2676–2685.
- Chen, J.; Stahl, A.; Krah, N.M.; Seaward, M.R.; Dennison, R.J.; Sapieha, P.; Hua, J.; Hatton, C.J.; Juan, A.M.; Aderman, C.M.; et al. Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation 2011, 124, 1871–1881.
- Chen, Q.; Ma, J. xing Canonical Wnt signaling in diabetic retinopathy. Vis. Res. 2017, 139, 47–58.
- Zhang, B.; Zhou, K.K.; Ma, J.X. Inhibition of connective tissue growth factor overexpression in diabetic retinopathy by SERPINA3K via blocking the WNT/β-catenin pathway. Diabetes 2010, 59, 1809–1816.
- Wang, Z.; Cheng, R.; Lee, K.; Tyagi, P.; Ding, L.; Kompella, U.B.; Chen, J.; Xu, X.; Ma, J.X. Nanoparticle-mediated expression of a Wnt pathway inhibitor ameliorates ocular neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 855–864.
- Takahashi, K.; Saishin, Y.; Saishin, Y.; Silva, R.L.; Oshima, Y.; Oshima, S.; Melia, M.; Paszkiet, B.; Zerby, D.; Kadan, M.J.; et al. Intraocular expression of endostatin reduces VEGF-induced retinal vascular permeability, neovascularization, and retinal detachment. FASEB J. 2003, 17, 896–898.
- Liu, X.; Zhang, B.; McBride, J.D.; Zhou, K.; Lee, K.; Zhou, Y.; Liu, Z.; Ma, J.X. Antiangiogenic and antineuroinflammatory effects of kallistatin through interactions with the canonical wnt pathway. Diabetes 2013, 62, 4228–4238.
- Park, K.; Lee, K.; Zhang, B.; Zhou, T.; He, X.; Gao, G.; Murray, A.R.; Ma, J.-X. Identification of a novel inhibitor of the canonical Wnt pathway. Mol. Cell. Biol. 2011, 31, 3038–3051.
- Takahashi, Y.; Chen, Q.; Rajala, R.V.S.; Ma, J.X. MicroRNA-184 modulates canonical Wnt signaling through the regulation of frizzled-7 expression in the retina with ischemia-induced neovascularization. FEBS Lett. 2015, 589, 1143–1149.
- Bose, M.; Almas, S.; Prabhakar, S. Wnt signaling and podocyte dysfunction in diabetic nephropathy. J. Investig. Med. 2017, 65, 1093–1101.
- Liu, Y.; Shang, D. Transforming growth factor-β1 enhances proliferative and metastatic potential by up-regulating lymphoid enhancer-binding factor 1/integrin αMβ2 in human renal cell carcinoma. Mol. Cell. Biochem. 2020, 465, 165–174.
- Rooney, B.; O’Donovan, H.; Gaffney, A.; Browne, M.; Faherty, N.; Curran, S.P.; Sadlier, D.; Godson, C.; Brazil, D.P.; Crean, J. CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: Implications for the pathogenesis of diabetic nephropathy. FEBS Lett. 2011, 585, 531–538.
- Kiewisz, J.; Skowronska, A.; Winiarska, A.; Pawlowska, A.; Kiezun, J.; Rozicka, A.; Perkowska-Ptasinska, A.; Kmiec, Z.; Stompor, T. WNT4 expression in primary and secondary kidney diseases: Dependence on staging. Kidney Blood Press. Res. 2019, 44, 200–210.
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86.
- Xiong, Y.; Zhou, L. The signaling of cellular senescence in diabetic nephropathy. Oxidative Med. Cell. Longev. 2019, 2019, 7495629.
- Xiao, L.; Wang, M.; Yang, S.; Liu, F.; Sun, L. A glimpse of the pathogenetic mechanisms of Wnt/β-catenin signaling in diabetic nephropathy. Biomed. Res. Int. 2013, 2013, 987064.
- Beaton, H.; Andrews, D.; Parsons, M.; Murphy, M.; Gaffney, A.; Kavanagh, D.; McKay, G.J.; Maxwell, A.P.; Taylor, C.T.; Cummins, E.P.; et al. Wnt6 regulates epithelial cell differentiation and is dysregulated in renal fibrosis. Am. J. Physiol. Ren. Physiol. 2016, 311, F35–F45.
- Resham, K.; Sharma, S.S. Pharmacologic inhibition of porcupine, disheveled, and β-catenin in Wnt signaling pathway ameliorates diabetic peripheral neuropathy in rats. J. Pain 2019, 20, 1338–1352.
- Itokazu, T.; Hayano, Y.; Takahashi, R.; Yamashita, T. Involvement of Wnt/β-catenin signaling in the development of neuropathic pain. Neurosci. Res. 2014, 79, 34–40.
- Vinik, A.; Flemmer, M. Diabetes and macrovascular disease. J. Diabetes Complicat. 2002, 16, 235–245.
- Isea, J.; Viloria, J.L.; Ponte, C.I.; Gómez, J.R. Complicaciones macrovasculares de la diabetes mellitus: Cardíacas, vásculo cerebrales y enfermedad arterial periférica. Rev. Venez. Endocrinol. Metab. 2012, 10, 96–110.
- Couffinhal, T.; Dufourcq, P. Common pathway between wnt and growth factor signaling in vascular smooth muscle cell proliferation? Circ. Res. 2006, 99, 1287–1289.
- Kun, D.; Grantham, R.N.; Trachte, A.L.; Mannion, J.D.; Wilson, C.L. Activation of the canonical Wnt/b -catenin pathway enhances monocyte adhesion to endothelial cells. Biochem. Biophys. Res. Commun. 2006, 347, 109–116.
- Ku, M.; Dejana, E. The role of Wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010, 107, 1798–1806.
- Novo-Rodríguez, C.; García-Fontana, B.; Luna-Del Castillo, J.D.D.; Andujar-Vera, F.; Avila-Rubio, V.; García-Fontana, C.; Morales-Santana, S.; Rozas-Moreno, P.; Munoz-Torres, M. Circulating levels of sclerostin are associated with cardiovascular mortality. PLoS ONE 2018, 13, e0199504.
- Morales-Santana, S.; Rozas-Moreno, P.; Antonio, J.; Ia-Salcedo, G.; Reyes-Garc Ia, R.; Muñoz-Torres, M. Atherosclerotic disease in type 2 diabetes is associated with an increase in sclerostin levels. Diabetes Care 2013, 36, 1667–1674.
- Ueland, T.; Åkerblom, A.; Ghukasyan, T.; Michelsen, A.E.; Becker, R.C.; Bertilsson, M.; Himmelmann, A.; James, S.K.; Siegbahn, A.; Storey, R.F.; et al. Admission levels of DKK1 (Dickkopf-1) are associated with future cardiovascular death in patients with acute coronary syndromes: Insights from the PLATO trial. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 294–302.
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577.
- Go, G.W.; Srivastava, R.; Hernandez-Ono, A.; Gang, G.; Smith, S.B.; Booth, C.J.; Ginsberg, H.N.; Mani, A. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell Metab. 2014, 19, 209–220.
- Relling, I.; Akcay, G.; Fangmann, D.; Knappe, C.; Schulte, D.M.; Hartmann, K.; Müller, N.; Türk, K.; Dempfle, A.; Franke, A.; et al. Role of Wnt5a in metabolic inflammation in humans. J. Clin. Endocrinol. Metab. 2018, 103, 4253–4264.
- Ohta, H.; Wada, H.; Niwa, T.; Kirii, H.; Iwamoto, N.; Fujii, H.; Saito, K.; Sekikawa, K.; Seishima, M. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 2005, 180, 11–17.
- Yu, B.; Kiechl, S.; Qi, D.; Wang, X.; Song, Y.; Weger, S.; Mayr, A.; Le Bras, A.; Karamariti, E.; Zhang, Z.; et al. A cytokine-like protein dickkopf-related protein 3 is atheroprotective. Circulation 2017, 136, 1022–1036.
- Uglow, E.B.; Slater, S.; Sala-Newby, G.B.; Aguilera-Garcia, C.M.; Angelini, G.D.; Newby, A.C.; George, S.J. Dismantling of cadherin-mediated cell-cell contacts modulates smooth muscle cell proliferation. Circ. Res. 2003, 92, 1314–1321.
- Srivastava, R.; Zhang, J.; Go, G.W.; Narayanan, A.; Nottoli, T.P.; Mani, A. Impaired LRP6-TCF7L2 activity enhances smooth muscle cell plasticity and causes coronary artery disease. Cell Rep. 2015, 13, 746–759.
- Williams, H.; Mill, C.A.E.; Monk, B.A.; Hulin-Curtis, S.; Johnson, J.L.; George, S.J. Wnt2 and WISP-1/CCN4 induce intimal thickening via promotion of smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1417–1424.
- Borrell-Pagès, M.; Romero, J.C.; Badimon, L. LRP5 deficiency down-regulates Wnt signalling and promotes aortic lipid infiltration in hypercholesterolaemic mice. J. Cell. Mol. Med. 2015, 19, 770–777.
- He, X.W.; Wang, E.; Bao, Y.Y.; Wang, F.; Zhu, M.; Hu, X.F.; Jin, X.P. High serum levels of sclerostin and Dickkopf-1 are associated with acute ischaemic stroke. Atherosclerosis 2016, 253, 22–28.
- Zhu, Z.; Guo, D.; Zhong, C.; Wang, A.; Xie, X.; Xu, T.; Chen, C.S.; Peng, Y.; Peng, H.; Li, Q.; et al. Serum Dkk-1 (Dickkopf-1) is a potential biomarker in the prediction of clinical outcomes among patients with acute ischemic stroke. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 285–293.
- Sun, J.D.; Li, X.M.; Liu, J.L.; Li, J.; Zhou, H. Effects of miR-150-5p on cerebral infarction rats by regulating the Wnt signaling pathway via p53. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3882–3891.
- Tian, W.; Zhu, W.; Jiang, J. miR-150-5p suppresses the stem cell-like characteristics of glioma cells by targeting the Wnt/β-catenin signaling pathway. Cell Biol. Int. 2020, 44, 1156–1167.
- Chong, Z.Z.; Shang, Y.C.; Hou, J.; Maiese, K. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxid. Med. Cell. Longev. 2010, 3, 153–165.
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; MacHado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.Y. Endothelial β-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation 2016, 133, 177–186.
- Yi, R.; Xiao-Ping, G.; Hui, L. Atorvastatin prevents angiotensin II-induced high permeability of human arterial endothelial cell monolayers via ROCK signaling pathway. Biochem. Biophys. Res. Commun. 2015, 459, 94–99.
- Ruan, W.; Hu, J.; Zhou, H.; Li, Y.; Xu, C.; Luo, Y.; Chen, T.; Xu, B.; Yan, F.; Chen, G. Intranasal Wnt-3a alleviates neuronal apoptosis in early brain injury post subarachnoid hemorrhage via the regulation of wnt target PPAN mediated by the moonlighting role of aldolase C. Neurochem. Int. 2020, 134, 104656.
- Abe, T.; Zhou, P.; Jackman, K.; Capone, C.; Casolla, B.; Hochrainer, K.; Kahles, T.; Ross, M.E.; Anrather, J.; Iadecola, C. Lipoprotein receptor-related protein-6 protects the brain from ischemic injury. Stroke 2013, 44, 2284–2291.
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via β-catenin–independent signaling. J. Immunol. 2010, 185, 1274–1282.
- Wang, J.; Chen, T.; Shan, G. MiR-148b regulates proliferation and differentiation of neural stem cells via Wnt/β-Catenin signaling in rat ischemic stroke model. Front. Cell. Neurosci. 2017, 11, 329.
- Wang, B.; Pan, Y.; Yang, G.; Cui, Z.; Yu, W.; Liu, H.; Bai, B. Sfrp5/Wnt5a and leptin/adiponectin levels in the serum and the periarterial adipose tissue of patients with peripheral arterial occlusive disease. Clin. Biochem. 2021, 87, 46–51.
- Myngbay, A.; Manarbek, L.; Ludbrook, S.; Kunz, J. The role of collagen triple helix repeat-containing 1 protein (Cthrc1) in rheumatoid arthritis. Int. J. Mol. Sci. 2021, 22, 2426.
- Zhao, Y.; Hu, J.; Sun, X.; Yang, K.; Yang, L.; Kong, L.; Zhang, B.; Li, F.; Li, C.; Shi, B.; et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin. Transl. Med. 2021, 11, e402.

