 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wiwin Effendi | -- | 1875 | 2022-06-30 23:55:37 | | | |
| 2 | Jessie Wu | Meta information modification | 1875 | 2022-07-01 03:19:01 | | | | |
| 3 | Jessie Wu | Meta information modification | 1875 | 2022-07-01 03:19:42 | | | | |
| 4 | Jessie Wu | Meta information modification | 1875 | 2022-07-04 08:35:48 | | |
Video Upload Options
Connective Tissue Growth Factor, also known as the cellular communication network 2 (CCN2), is a TGF-β-target gene and a member of the CCN family of secreted proteins that regulate matricellular protein. Matricellular proteins are expressed at higher levels during physiological and pathological processes, with distinct functions that bind to multiple receptors, other growth factors, and proteases, modulating their activity and mediating cross-talk between the ECM and cells. Idiopathic Pulmonary Fibrosis is a chronic, devastating, irreversible lung disease, characterized by injury-induced alveolar epithelial cell stress, progressive pathogenic myofibroblast differentiation, and imbalanced macrophage polarization, resulting in ECM deposition.
1. Structure, Regulation, and Function of Connective Tissue Growth Factor
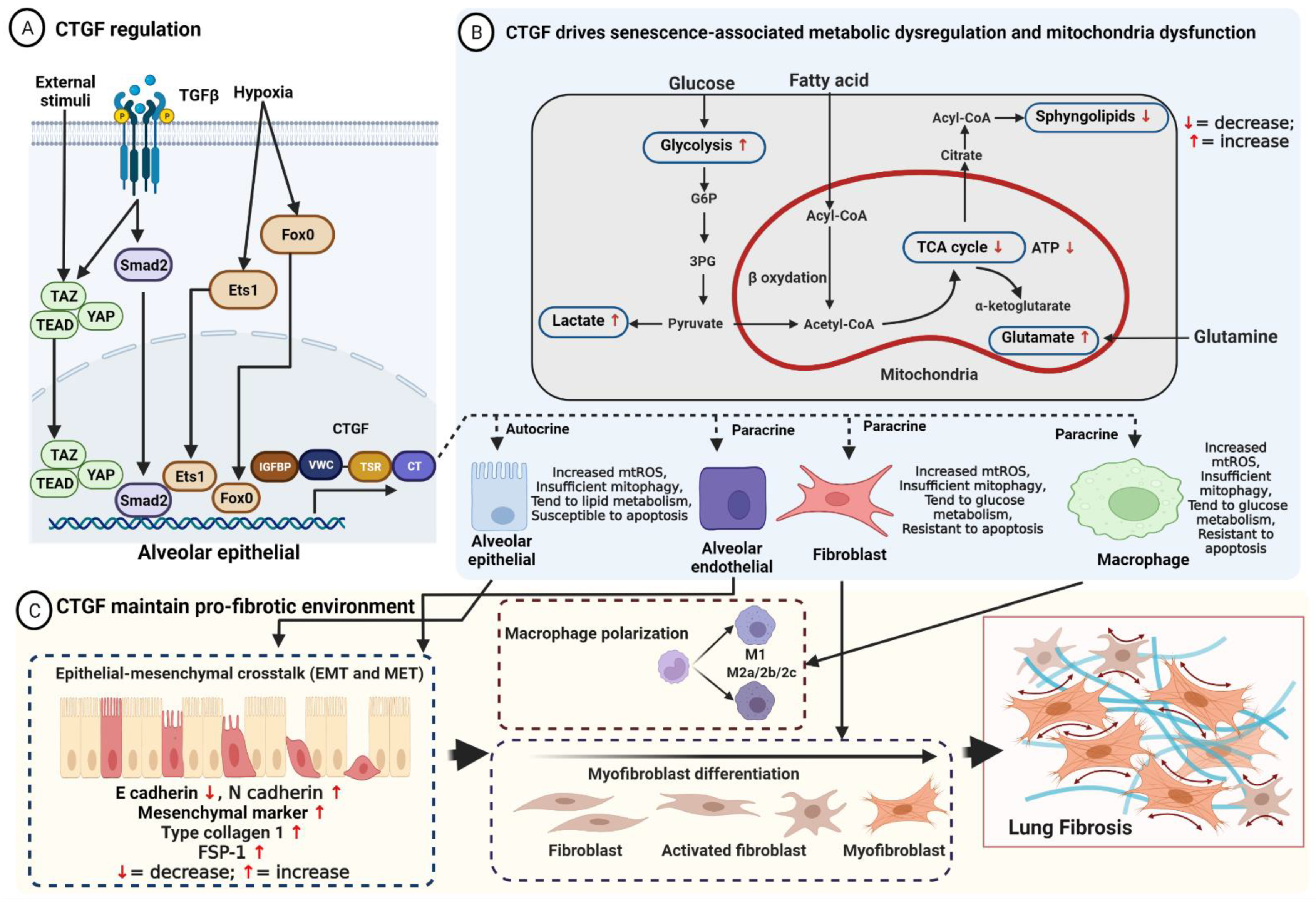
2. Connective Tissue Growth Factor Maintains the Pro-Fibrotic Environment in Idiopathic Pulmonary Fibrosis
2.1. Activated Alveolar Epithelial Cells Initiate a Cycle of Fibrosis through Connective Tissue Growth Factor
2.2. Connective Tissue Growth Factor Stimulates the Differentiation of Lung Fibroblasts
2.3. Connective Tissue Growth Factor Modulates Dysfunction of Macrophage Polarization
2.4. Connective Tissue Growth Factor Increases Endothelial Growth
2.5. Fibrocyte Differentiation Involved in Connective Tissue Growth Factor
References
- Holbourn, K.P.; Acharya, K.R.; Perbal, B. The CCN family of proteins: Structure-function relationships. Trends Biochem. Sci. 2008, 33, 461–473.
- Jun, J.-I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963.
- Shi-Wen, X.; Leask, A.; Abraham, D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008, 19, 133–144.
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008, 28, 1065–1079.
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. 2014, 128, 181–196.
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66.
- Preisser, F.; Giehl, K.; Rehm, M.; Goppelt-Struebe, M. Inhibitors of oxygen sensing prolyl hydroxylases regulate nuclear localization of the transcription factors Smad2 and YAP/TAZ involved in CTGF synthesis. Biochim. Biophys. Acta 2016, 1863, 2027–2036.
- Van Beek, J.P.; Kennedy, L.; Rockel, J.S.; Bernier, S.M.; Leask, A. The induction of CCN2 by TGFbeta1 involves Ets-1. Arthritis Res. Ther. 2006, 8, R36.
- Samarin, J.; Cicha, I.; Goppelt-Struebe, M. Cell type-specific regulation of CCN2 protein expression by PI3K-AKT-FoxO signaling. J. Cell Commun. Signal. 2009, 3, 79–84.
- Liao, X.; Bu, Y.; Jiang, S.; Chang, F.; Jia, F.; Xiao, X.; Song, G.; Zhang, M.; Ning, P.; Jia, Q. CCN2-MAPK-Id-1 loop feedback amplification is involved in maintaining stemness in oxaliplatin-resistant hepatocellular carcinoma. Hepatol. Int. 2019, 13, 440–453.
- Ihn, H. Pathogenesis of fibrosis: Role of TGF-beta and CTGF. Curr. Opin. Rheumatol. 2002, 14, 681–685.
- Yeger, H.; Perbal, B. CCN family of proteins: Critical modulators of the tumor cell microenvironment. J. Cell Commun. Signal. 2016, 10, 229–240.
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.F.; Goldschmeding, R.; Schlingemann, R.O. The Angio-Fibrotic Switch of VEGF and CTGF in Proliferative Diabetic Retinopathy. PLoS ONE 2008, 3, e2675.
- Zhao, Z.; Ho, L.; Wang, J.; Qin, W.; Festa, E.D.; Mobbs, C.; Hof, P.; Rocher, A.; Masur, S.; Haroutunian, V.; et al. Connective tissue growth factor (CTGF) expression in the brain is a downstream effector of insulin resistance- associated promotion of Alzheimer’s disease beta-amyloid neuropathology. FASEB J. 2005, 19, 2081–2082.
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-β1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209.
- Makino, K.; Makino, T.; Stawski, L.; Lipson, K.E.; Leask, A.; Trojanowska, M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res. Ther. 2017, 19, 134.
- Yan, L.; Chaqour, B. Cysteine-rich protein 61 (CCN1) and connective tissue growth factor (CCN2) at the crosshairs of ocular neovascular and fibrovascular disease therapy. J. Cell Commun. Signal. 2013, 7, 253.
- Hou, N.; Wen, Y.; Yuan, X.; Xu, H.; Wang, X.; Li, F.; Ye, B. Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Exp. Mol. Pathol. 2017, 103, 267–275.
- Accornero, F.; van Berlo, J.H.; Correll, R.N.; Elrod, J.W.; Sargent, M.A.; York, A.; Rabinowitz, J.E.; Leask, A.; Molkentin, J.D. Genetic Analysis of Connective Tissue Growth Factor as an Effector of Transforming Growth Factor β Signaling and Cardiac Remodeling. Mol. Cell. Biol. 2015, 35, 2154–2164.
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641.
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; Ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719.
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways. Int. J. Mol. Sci. 2016, 17, 2091.
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151.
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823.
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129.
- Kage, H.; Borok, Z. EMT and Interstitial Lung Disease: A Mysterious Relationship. Curr. Opin. Pulm. Med. 2012, 18, 517.
- Kolb, M.; Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers—Insights from the Bench Side. Respiration 2013, 86, 441–452.
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483.
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638.
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096.
- Xu, X.; Dai, H.; Wang, C. Epithelium-dependent profibrotic milieu in the pathogenesis of idiopathic pulmonary fibrosis: Current status and future directions. Clin. Respir. J. 2016, 10, 133–141.
- Rabeyrin, M.; Thivolet, F.; Ferretti, G.R.; Chalabreysse, L.; Jankowski, A.; Cottin, V.; Pison, C.; Cordier, J.-F.; Lantuejoul, S. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann. Diagn. Pathol. 2015, 19, 269–276.
- Vanstapel, A.; Goldschmeding, R.; Broekhuizen, R.; Nguyen, T.; Sacreas, A.; Kaes, J.; Heigl, T.; Verleden, S.E.; De Zutter, A.; Verleden, G.; et al. Connective Tissue Growth Factor Is Overexpressed in Explant Lung Tissue and Broncho-Alveolar Lavage in Transplant-Related Pulmonary Fibrosis. Front. Immunol. 2021, 12, 661761.
- Ponticos, M.; Holmes, A.M.; Shi-wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009, 60, 2142–2155.
- Xu, X.; Wan, X.; Geng, J.; Li, F.; Yang, T.; Dai, H. Rapamycin regulates connective tissue growth factor expression of lung epithelial cells via phosphoinositide 3-kinase. Exp. Biol. Med. 2013, 238, 1082–1094.
- Kono, M.; Nakamura, Y.; Suda, T.; Kato, M.; Kaida, Y.; Hashimoto, D.; Inui, N.; Hamada, E.; Miyazaki, O.; Kurashita, S.; et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin. Chim. Acta 2011, 412, 2211–2215.
- Yanagihara, T.; Tsubouchi, K.; Gholiof, M.; Chong, S.G.; Lipson, K.E.; Zhou, Q.; Scallan, C.; Upagupta, C.; Tikkanen, J.; Keshavjee, S.; et al. Connective-Tissue Growth Factor Contributes to TGF-β1-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 260–270.
- Morishima, Y.; Nomura, A.; Uchida, Y.; Noguchi, Y.; Sakamoto, T.; Ishii, Y.; Goto, Y.; Masuyama, K.; Zhang, M.J.; Hirano, K.; et al. Triggering the induction of myofibroblast and fibrogenesis by airway epithelial shedding. Am. J. Respir. Cell Mol. Biol. 2001, 24, 1–11.
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957.
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227.
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L786–L796.
- Zhou, T.; Yu, Q.; Lin, H.; Wang, Z.; Fu, G.; Lei, L.; Shi, Y.; Zhang, L.; Qin, L.; Liu, Y. The Role of CTGF in Inflammatory Responses Induced by Silica Particles in Human Bronchial Epithelial Cells. Lung 2019, 197, 783–791.
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56.
- Shi, L.; Dong, N.; Fang, X.; Wang, X. Regulatory mechanisms of TGF-β1-induced fibrogenesis of human alveolar epithelial cells. J. Cell. Mol. Med. 2016, 20, 2183–2193.
- Sonnylal, S.; Xu, S.; Jones, H.; Tam, A.; Sreeram, V.R.; Ponticos, M.; Norman, J.; Agrawal, P.; Abraham, D.; de Crombrugghe, B. Connective tissue growth factor causes EMT-like cell fate changes in vivo and in vitro. J. Cell Sci. 2013, 126, 2164–2175.
- Shafieian, M.; Chen, S.; Wu, S. Integrin-linked kinase mediates CTGF-induced epithelial to mesenchymal transition in alveolar type II epithelial cells. Pediatr. Res. 2015, 77, 520–527.
- Cheng, Y.; Lin, C.H.; Chen, J.Y.; Li, C.H.; Liu, Y.T.; Chen, B.C. Induction of Connective Tissue Growth Factor Expression by Hypoxia in Human Lung Fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 Pathways. PLoS ONE 2016, 11, e0160593.
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610.
- Ou, S.-C.; Bai, K.-J.; Cheng, W.-H.; Chen, J.-Y.; Lin, C.-H.; Wen, H.-C.; Chen, B.-C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int. J. Mol. Sci. 2020, 21, 9084.
- Madala, S.K.; Schmidt, S.; Davidson, C.; Ikegami, M.; Wert, S.; Hardie, W.D. MEK-ERK Pathway Modulation Ameliorates Pulmonary Fibrosis Associated with Epidermal Growth Factor Receptor Activation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 380–388.
- Zhang, C.; Meng, X.; Zhu, Z.; Liu, J.; Deng, A. Connective tissue growth factor regulates the key events in tubular epithelial to myofibroblast transition in vitro. Cell Biol. Int. 2004, 28, 863–873.
- Gore-Hyer, E.; Shegogue, D.; Markiewicz, M.; Lo, S.; Hazen-Martin, D.; Greene, E.L.; Grotendorst, G.; Trojanowska, M. TGF-β and CTGF have overlapping and distinct fibrogenic effects on human renal cells. Am. J. Physiol. Physiol. 2002, 283, F707–F716.
- Hung, C.F. Origin of Myofibroblasts in Lung Fibrosis. Curr. Tissue Microenviron. Rep. 2020, 1, 155–162.
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208.
- Garrett, Q.; Khaw, P.T.; Blalock, T.D.; Schultz, G.S.; Grotendorst, G.R.; Daniels, J.T. Involvement of CTGF in TGF-beta1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1109–1116.
- Folger, P.A.; Zekaria, D.; Grotendorst, G.; Masur, S.K. Transforming growth factor-beta-stimulated connective tissue growth factor expression during corneal myofibroblast differentiation. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2534–2541.
- Yang, Z.; Sun, Z.; Liu, H.; Ren, Y.; Shao, D.; Zhang, W.; Lin, J.; Wolfram, J.; Wang, F.; Nie, S. Connective tissue growth factor stimulates the proliferation, migration and differentiation of lung fibroblasts during paraquat-induced pulmonary fibrosis. Mol. Med. Rep. 2015, 12, 1091–1097.
- Tsai, C.-C.; Wu, S.-B.; Kau, H.-C.; Wei, Y.-H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276.
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45.
- Tam, A.Y.Y.; Horwell, A.L.; Trinder, S.L.; Khan, K.; Xu, S.; Ong, V.; Denton, C.P.; Norman, J.T.; Holmes, A.M.; Bou-Gharios, G.; et al. Selective deletion of connective tissue growth factor attenuates experimentally-induced pulmonary fibrosis and pulmonary arterial hypertension. Int. J. Biochem. Cell Biol. 2021, 134, 105961.
- Piera-Velazquez, S.; Jimenez, S.A. Oxidative Stress Induced by Reactive Oxygen Species (ROS) and NADPH Oxidase 4 (NOX4) in the Pathogenesis of the Fibrotic Process in Systemic Sclerosis: A Promising Therapeutic Target. J. Clin. Med. 2021, 10, 4791.
- Shibata, S.; Ishiyama, J. Secreted protein acidic and rich in cysteine (SPARC) is upregulated by transforming growth factor (TGF)-β and is required for TGF-β-induced hydrogen peroxide production in fibroblasts. Fibrogenesis Tissue Repair 2013, 6, 6.
- Wang, J.-C.; Lai, S.; Guo, X.; Zhang, X.; de Crombrugghe, B.; Sonnylal, S.; Arnett, F.C.; Zhou, X. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res. Ther. 2010, 12, R60.
- Wang, J.C.; Sonnylal, S.; Arnett, F.C.; De Crombrugghe, B.; Zhou, X. Attenuation of expression of extracellular matrix genes with siRNAs to Sparc and Ctgf in skin fibroblasts of CTGF transgenic mice. Int. J. Immunopathol. Pharmacol. 2011, 24, 595–601.
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170.
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457.
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Dunn, J.C.; Bolus, W.R.; Kendall, P.L.; Hasty, A.H.; Gannon, M. Macrophages are essential for CTGF-mediated adult β-cell proliferation after injury. Mol. Metab. 2015, 4, 584–591.
- Wang, T.-T.; Yuan, J.-H.; Ma, J.-Z.; Yang, W.-J.; Liu, X.-N.; Yin, Y.-P.; Liu, Y.; Pan, W.; Sun, S.-H. CTGF secreted by mesenchymal-like hepatocellular carcinoma cells plays a role in the polarization of macrophages in hepatocellular carcinoma progression. Biomed. Pharmacother. 2017, 95, 111–119.
- Zhang, S.-M.; Wei, C.-Y.; Wang, Q.; Wang, L.; Lu, L.; Qi, F.-Z. M2-polarized macrophages mediate wound healing by regulating connective tissue growth factor via AKT, ERK1/2, and STAT3 signaling pathways. Mol. Biol. Rep. 2021, 48, 6443–6456.
- Bickelhaupt, S.; Erbel, C.; Timke, C.; Wirkner, U.; Dadrich, M.; Flechsig, P.; Tietz, A.; Pföhler, J.; Gross, W.; Peschke, P.; et al. Effects of CTGF Blockade on Attenuation and Reversal of Radiation-Induced Pulmonary Fibrosis. J. Natl. Cancer Inst. 2017, 109, djw339.
- Li, Z.; Jimenez, S.A. Protein kinase Cδ and c-Abl kinase are required for transforming growth factor β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011, 63, 2473–2483.
- Martin, M.; Zhang, J.; Miao, Y.; He, M.; Kang, J.; Huang, H.-Y.; Chou, C.-H.; Huang, T.-S.; Hong, H.-C.; Su, S.-H.; et al. Role of endothelial cells in pulmonary fibrosis via SREBP2 activation. JCI Insight 2021, 6, e125635.
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327.
- Brigstock, D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002, 5, 153–165.
- Pi, L.; Fu, C.; Lu, Y.; Zhou, J.; Jorgensen, M.; Shenoy, V.; Lipson, K.E.; Scott, E.W.; Bryant, A.J. Vascular Endothelial Cell-Specific Connective Tissue Growth Factor (CTGF) Is Necessary for Development of Chronic Hypoxia-Induced Pulmonary Hypertension. Front. Physiol. 2018, 9, 138.
- Liu, S.-C.; Chuang, S.-M.; Hsu, C.-J.; Tsai, C.-H.; Wang, S.-W.; Tang, C.-H. CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis. 2014, 5, e1485.
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863.
- Kato, S.; Inui, N.; Hakamata, A.; Suzuki, Y.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; Watanabe, H.; Suda, T. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 2018, 19, 127.
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; van den Toorn, L.M.; Boomars, K.A.T.; Wijsenbeek, M.S.; Hoogsteden, H.; von der Thüsen, J.H.; et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 90.
- Rosin, N.L.; Falkenham, A.; Sopel, M.J.; Lee, T.D.G.; Légaré, J.-F. Regulation and Role of Connective Tissue Growth Factor in AngII-Induced Myocardial Fibrosis. Am. J. Pathol. 2013, 182, 714–726.
- Weng, C.-M.; Chen, B.-C.; Wang, C.-H.; Feng, P.-H.; Lee, M.-J.; Huang, C.-D.; Kuo, H.-P.; Lin, C.-H. The Endothelin A Receptor Mediates Fibrocyte Differentiation in Chronic Obstructive Asthma. The Involvement of Connective Tissue Growth Factor. Am. J. Respir. Crit. Care Med. 2013, 188, 298–308.
- Wang, T.-Y.; Lo, Y.-L.; Wang, C.-H.; Kuo, H.-P. Increased CTGF expression of circulating fibrocytes in asthmatic patients with severe OSA—The role of HIF-1a and HDAC7. Eur. Respir. J. 2018, 52, PA2186.


