Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Clelia Donisi | -- | 2689 | 2022-06-30 23:15:10 | | | |
| 2 | Jessie Wu | Meta information modification | 2689 | 2022-07-01 03:35:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Madeddu, C.; Donisi, C.; Liscia, N.; Lai, E.; Scartozzi, M.; Macciò, A. Role of Tumor Microenvironment and EGFR Mutations. Encyclopedia. Available online: https://encyclopedia.pub/entry/24709 (accessed on 27 June 2026).
Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Macciò A. Role of Tumor Microenvironment and EGFR Mutations. Encyclopedia. Available at: https://encyclopedia.pub/entry/24709. Accessed June 27, 2026.
Madeddu, Clelia, Clelia Donisi, Nicole Liscia, Eleonora Lai, Mario Scartozzi, Antonio Macciò. "Role of Tumor Microenvironment and EGFR Mutations" Encyclopedia, https://encyclopedia.pub/entry/24709 (accessed June 27, 2026).
Madeddu, C., Donisi, C., Liscia, N., Lai, E., Scartozzi, M., & Macciò, A. (2022, June 30). Role of Tumor Microenvironment and EGFR Mutations. In Encyclopedia. https://encyclopedia.pub/entry/24709
Madeddu, Clelia, et al. "Role of Tumor Microenvironment and EGFR Mutations." Encyclopedia. Web. 30 June, 2022.
Copy Citation
Lung cancer is a leading cause of cancer-related deaths worldwide. About 10–30% of patients with non-small cell lung cancer (NSCLC) harbor mutations of the EGFR gene. The Tumor Microenvironment (TME) of patients with NSCLC harboring EGFR mutations displays peculiar characteristics and may modulate the antitumor immune response. EGFR activation increases PD-L1 expression in tumor cells, inducing T cell apoptosis and immune escape. EGFR activation increases PD-L1 expression in tumor cells, inducing T cell apoptosis and immune escape.
EGFR
tumor microenvironment
tumor-associated macrophages
1. Role of Tumor-Associated Macrophages and Related Inflammation on the Efficacy of Immune Response and Immunotherapy
Tumor-associated macrophages (TAMs) are essential elements during the initial phase of the immune response due to their phagocytic capacity, ability to synthesize interferon (IFN), and interactions with helper and cytotoxic T lymphocytes. However, their persistent activation with consequent chronicization of inflammation, oxidative stress, and changes in metabolic pathways lead to the impairment of effective T-cell responses by causing T-cell exhaustion, a condition in which lymphocytes, even when activated, are non-functional and subsequently undergo programmed cell death [1]. In this regard, Mascaux et al. [2] demonstrated that in lung squamous cell carcinoma, the adaptive immune response was the strongest at the earliest cancer stages, whereas at the most advanced invasive stages, they observed increased expression levels of co-inhibitory molecules and suppressive cytokines, such as PD-L1, IL-10, and IL-6.
To understand the role of TAMs in the efficacy of immune response and immunotherapy, it is necessary to clarify the contribution of Tregs to cancer immunosuppression. Tregs are a specialized T-cell lineage that express the transcription factor FOXP3, which is crucial for Treg stabilization and stimulation of the Treg-specific gene expression profile necessary to prevent autoimmune reactions in normal conditions [3][4]. However, Tregs can switch their fate and phenotype under certain circumstances, such as inflammatory perturbations of the microenvironment. This is possible owing to changes in their gene expression program, which is characterized by the loss of FOXP3 expression and production of pro-inflammatory cytokines, and IFN-γ, which convert these cells into effector T-cells (Treg reprogramming). In a recent study by Di Pilato et al., an increase in IFN-γ levels favored the expression of PD-1 on effector T-cells and synthesis of PD-L1 by cancer cells and macrophages [5], thereby turning off CD4+CD25− conventional effector T-cells, reactivating an immune escape mechanism and increasing macrophage activation [6]. Recently, Gallimore et al. highlighted an important therapeutic role of the induction of selective recruitment and modulation of different Tregs with different molecular profiles and functions in the TME [7]. Based on these observations, other authors observed that immunotherapy efficacy could be diminished by the inflammatory response caused by the reprogramming of Tregs [1]. Indeed, as already described above, various suppressive and counter-regulatory mechanisms may be involved in the lack of immunotherapy effectiveness, especially in the advanced stages of neoplastic disease. In patients with advanced cancer, specific changes in oxidative and glycolytic metabolic pathways during a chronic inflammatory response interfere with conventional T-cell activation and function and may be one of the reasons for the ineffectiveness of immunotherapy. Consequently, a combined strategy of modulating the activity of Tregs, pharmacological inhibition of chronic inflammation mediated by macrophages, and, at the same time, suppression of oxidative stress and positive regulation of metabolic imbalances could improve the effectiveness of modern immunotherapy. Reprogramming of Tregs has a dual effect: firstly, it immediately activates immunosurveillance, and secondly, it causes delayed, macrophage-mediated inflammation deleterious for the antineoplastic efficiency of the immune system response [8]. Furthermore, the loss of Treg activity shown in various in vivo and in vitro experimental models involving reprogramming of Tregs into IFN-γ-producing cells [9][10][11], is accompanied by M1 pro-inflammatory polarization of peritoneal macrophages with associated production of pro-inflammatory and immunosuppressive cytokines [12]. The persistent activation of macrophages does not favor sustained antitumor T-cell responses [13][14]. Following M1 macrophage polarization, other processes such as increased synthesis proinflammatory cytokines, production of reactive oxygen species, and changes in glucose and iron metabolism occur [15]. In particular, an iron-sequestering phenotype develops, which is characterized by intracellular iron accumulation and low iron release and availability (functional iron deficiency) [16]. Dysregulation of the iron metabolism impairs several vital cell processes, such as DNA and protein synthesis, enzyme activity, integrity of oxidative pathways, and cell proliferation, thereby resulting in a progressive loss of T-cell function when cancer advances [17].
Based on this evidence, a strategy that blocks chronic inflammation and Treg reprogramming should be considered in some patients depending on the cancer stage. Moreover, TAMs, specifically M1, are the main producers of IL-6, which plays a key role in modulating both tumor progression and immune escape through multiple mechanisms [1][18]. In particular, IL-6 is involved in lung cancer tumorigenesis, and its increased circulating levels have been associated with poor survival of patients with lung cancer [19]. In addition, IL-6 acts directly on lung epithelial cells via the nuclear factor kappa B signaling pathway when conditioned by exposure to carcinogens. Tobacco smoking is known to induce KRAS mutations and thereby stimulate IL-6 expression in the lung epithelium [20], promoting lung cancer cell proliferation and migration through the STAT3 pathway activation [21]. Exhausted tumor-associated CD8+ T lymphocytes are another source of IL-6 in lung cancer [22]. Moreover, IL-6 is one of the key cytokines driving the immunopathology caused by the prolonged non-specific inflammation contributing to the so-called “cytokine storm”, with related systemic symptoms and impairment of immune response. Indeed, IL-6 influences the effectiveness of the immune system in multiple ways. IL-6 can act as an activator or inhibitor of T-cell responses, depending on the duration of its activity; moreover, by inducing systemically specific derangements of energy metabolism, nutritional status, and symptoms as anemia and anorexia, it significantly negatively affects T-cell functions [23]. Elevated levels of IL-6 are often observed in advanced lung cancer patients, which, at diagnosis, frequently present cachexia syndrome, an inflammatory driven severe condition characterized by involuntary weight-loss accompanied by chronic inflammation, fatigue, anorexia, and anemia, where IL-6 is actually one of the key pathogenetic mediators [23]. Thus, although IL-6 initially participates in the activation of the immune response, its prolonged, chronic release ultimately contributes to immunosuppression, severe cancer-related symptoms, and poor general patient status and prognosis. Consistent with the above evidence, blocking chronic inflammation, primarily driven by IL-6, may be fundamental in improving the efficacy of currently available immunotherapy, especially in advanced lung cancer patients [1].
2. Role of EGFR Mutations in Influencing Tumor Microenvironment, Tumor-Associated Macrophage Polarization, and Response to Immunotherapy
Preclinical and clinical studies have pointed out that the TME of patients with NSCLC harboring EGFR mutations displays peculiar characteristics and may modulate the antitumor immune response [24]. Several trials indicated that EGFR mutations are associated with immunosuppressive TME, lower tumor mutation burden (TMB), and increased PD-L1 expression [24][25]. TMB is defined as the total number of substitution, insertion, and deletion mutations per megabase of the coding region that encodes a tumor gene. Recent studies suggested that reduced TMB may predict a poor response to immune checkpoint inhibitors (ICIs) in patients carrying EGFR mutations [24][26]. Preclinical studies indicated that EGFR mutations lead to cancer immune escape through the PD-1/PD-L1 pathway [24]. In addition, it has been shown that EGFR mutations influence TME components, such as tumor-infiltrating lymphocytes (TILs), Tregs, MDSCs, TAMs, and immunoregulatory cytokines (Figure 1).
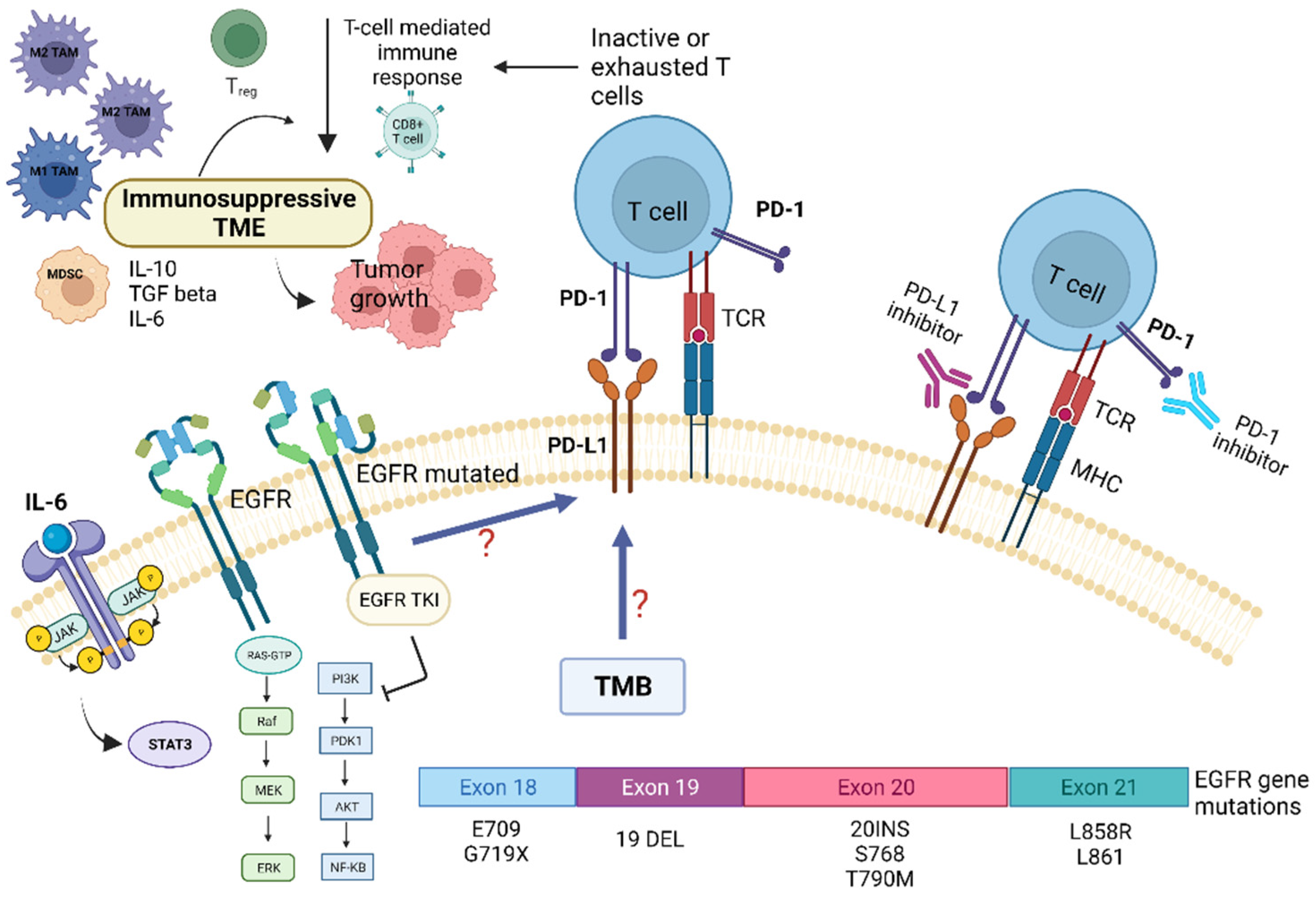
Figure 1. Role of tumor microenvironment in EGFR-mutated NSCLC in influencing resistance pathways to targeted TKI treatment and potential targets for immunotherapy. EGFR mutations are associated with immunosuppressive TME, lower tumor-mutation burden (TMB), and increased PD-L1 expression. EGFR mutations may promote cancer immune escape through modulation of the PD-1/PD-L1 pathway, which in turn determine T-cells inactivity and/or exhaustion. This also leads to EGFR-TKI resistance. In addition, EGFR mutations influence several TME components, such as tumor-infiltrating lymphocytes (TILs), Tregs, MDSCs, TAMs, and immunoregulatory/proinflammatory cytokines, i.e., IL-6. The latter, through the activation of the STAT-3 intracellular pathway, contribute to tumor growth and resistance to targeted therapies. Abbreviations: AKT—serine-threonine kinase; EGFR, epidermal growth factor receptor; ERK—extracellular signal-regulated kinase; IL—Interleukin; JAK—Janus kinase; MHC—major histocompatibility complex; MEK—mitogen-activated protein kinase; MDSC—myeloid-derived suppressor cells; NF-kB, nuclear factor kappa B; PI3K—phosphatidylinositol-4,5-bisphosphate 3-kinase; PD-1—programmed death; PD-L1—programmed death ligand-1; TKI—Tyrosine kinase inhibitors; Treg—regulatory T-cell; STAT3—signal transducer and activator of transcription 3; TCR—T-cell receptor; TMB—tumor mutational burden. Created with BioRender.com (https://biorender.com/, accessed on 17 May 2022).
A retrospective study observed that NSCLC tumors harboring EGFR mutations had low expression levels of PD-L1 and few CD8+ TILs. In contrast, other studies have detected high PD-L1 expression in this type of tumor [27]. Preclinical studies have demonstrated that EGFR activation upregulates intrinsic PD-L1 expression, inducing T-cell apoptosis and immune escape in EGFR-mutated NSCLC. In a genetically engineered mouse model of lung adenocarcinoma carrying an EGFR mutation, decreased macrophage MHC-II expression, enhanced macrophage IL1RA expression, and increased macrophage phagocytic activity have been observed and attributed to the M2 macrophage phenotype [28]. The presence of inflamed TME is considered a positive predictive factor for the response to immunotherapy. Although EGFR-mutated NSCLC typically is not associated with inflamed TME, characterized by low levels of CD8+ T cells and immune-suppressive cells, the numbers of Tregs and PD-L1 expression levels are increased in this cancer (Figure 1).
This leads to reduced effector T-cell activity and promotes TME favoring immune escape and cancer progression [28][29]. In EGFR-mutated cancers, TME displays high Treg infiltration without CD8+ T-cell infiltration. The recruitment of effector CD8+ T cells is prevented by the downregulation of CXCL10 through IRF1. In contrast, the stimulation of Treg infiltration is achieved through the upregulation of CCL22 through JNK/-JUN. Such immunological status may correlate with resistance to immunotherapy. Moreover, in EGFR-mutated cancers, the non-inflamed immunosuppressive TME (high levels of Tregs and low levels of CD8+ T cells) diminishes the expression of EGFR by Tregs, leading to the development of the resistance to TKIs. In conclusion, in the TME of EGFR-mutated NSCLC, high Treg infiltration occurs in the context of the non-inflamed TME. Therefore, EGFR mutations play a crucial role in cell growth, survival, and development of immune escape mechanisms [28].
3. Dynamic Changes of the Tumor Microenvironment during Tyrosine Kinase Inhibitor Treatment
Treatment with EGFR-TKIs alters the TME and decreases PD-L1 expression levels, which may also affect the response to immunotherapy. Additionally, EGFR-TKIs regulate the strength of the immune response through TME changes (Figure 2). In particular, EGFR-TKIs increase the presentation of MHC class I and II molecules and potentiate T-cell-mediated tumor killing. Moreover, the numbers of tumor-infiltrating effector Tregs were significantly lower in patients treated with TKIs. The lung cancer TME contains CD8+ T cells and immune-suppressive TAMs expressing PD-L1. From a clinical standpoint, the presence of PD-L1+ TAMs may predict the effectiveness of ICIs.
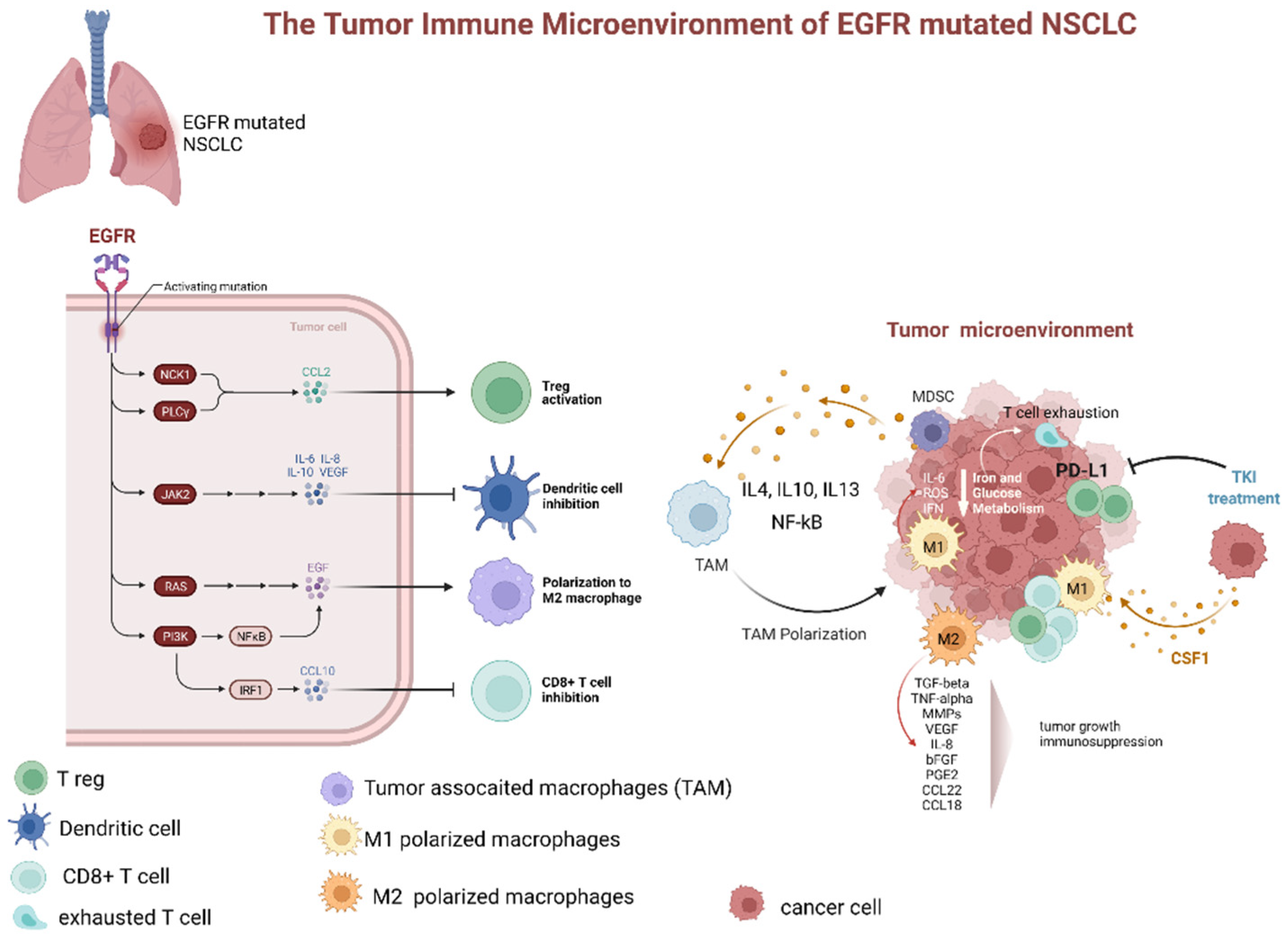
Figure 2. Dynamic changes of tumor microenvironment (TME) of EGFR-mutated NSCLC during tyrosine kinase inhibitor treatment. TME of EGFR mutated adenocarcinoma is typically characterized by prevalence of M2 polarized macrophage, low levels of CD8+ cells, increased number of Treg, and upregulation of PD-L1. The latter, especially if associated with macrophage-mediated inflammation particularly through IL-6 and increased ROS levels, contributes to T-cell exhaustion. Additionally, several factors secreted by M2 polarized TAMs (as TGFbeta, TNFalpha, MMPs, VEGF, IL-8, bFGF, PGE2, CCL22, and CCL18). These factors contribute to tumor progression and immunodepression. The TKI treatment has been associated with a decrease in PD-L1 expression, lowering of Treg, and promotion of TAM polarization from M2 to M1 phenotype. Abbreviations: EGFR—epidermal growth factor receptor; NSCLC—non-small cell lung cancer; JAK—Janus Kinase; PI3K—phosphatidylinositol-3 kinase; NF-kB—nuclear factor-κB; IRF1—interferon regulatory factor 1; IL—interleukin; TAM—tumor-associated macrophages; MDCS—myeloid-derived suppressor cells; PD-L1—programmed death-ligand 1; ROS—reactive oxygen species; IFN—interferon; TKI—tyrosine kinase inhibitor; CSF1—colony stimulating factor 1; TGF—tumor growth factor; TNF—tumor necrosis factor; MMP—matrix metalloproteases; VEGF—vascular endothelial growth factor; PGE—prostaglandin E; CCL—C-C-motif ligand. Created with BioRender.com (https://biorender.com/, accessed on 17 May 2022).
Although according to one clinical trial, pembrolizumab did not elicit a significant response in patients with EGFR-mutated lung cancer naïve for EGFR-TKI, even in the presence of high PD-L1 expression, the efficacy of this ICI could be influenced by TME changes during the EGFR-TKI treatment [30][31]. Recently, IT effectiveness was shown to correlate positively with the number of CD8+ lymphocytes and negatively with the number of FOXP3+ tumor-infiltrating lymphocytes in patients who acquired resistance to EGFR-TKIs [32]. In another study, both mouse and human macrophages were demonstrated to prevent killing of cancer cells by CD8+ T-cells, thereby affecting the response to immunotherapy [33]. Nonetheless, according to lung cancer clinical data from case series and clinical trials, the presence of TAMs expressing PD-L1 apparently correlates with a good response to immunotherapy, owing to the negative effects on cytotoxic lymphocytes [34][35][36][37]. A retrospective study evaluated the effectiveness of immunotherapy in patients with EGFR-mutated NSCLC by assessing both PD-L1 expression and TME parameters, including numbers of CD8+ TILs [38]. On the basis of PD-L1 expression levels and abundance of CD8+ TILs, the TME was divided into four subtypes: type I—adaptive immune resistance (PD-L1+/CD8+); type II—immune ignorance (PD-L1−/CD8−); type III—intrinsic induction (PD-L1+/CD8−); and type IV—immune tolerance (PD-L1−/CD8+) [39]. The results of that study confirmed that TKIs alter PD-L1 and PD-L2 expression levels and affect the numbers of CD8+ TILs. High abundance of CD8+ TILs was shown to be associated with favorable outcomes in EGFR-mutated NSCLC. Furthermore, high levels of CD8+ TILs may affect the response to EGFR-TKI treatment [38]. Su et al. [40] reported a high proportion of PD-L1+/CD8+ cases among patients with de novo resistance to first-line EGFR-TKIs for advanced NSCLC. These findings suggested that NSCLCs with high PD-L1 expression and large numbers of CD8+ TILs benefit less from TKI treatment despite EGFR mutations. In addition, many reports indicated that relatively high PD-L1 expression in EGFR-mutated NSCLC was related to lower response to EGFR-TKIs and worse progression-free survival [38][41][42].
In recent clinical trials, it has been observed that ICIs promote macrophage polarization from the M2 to M1 phenotype [34]. In a retrospective study evaluating the relationship between TAMs and response to EGFR-TKIs in treatment-naïve patients, irrespective of the EGFR mutation status, both univariate and multivariate analyses showed that TAMs and EGFR mutations were independent prognostic factors of survival. However, as proposed in the review by Biswas et al. [43], these parameters correlate with each other. Tumors carrying EGFR mutations had higher TAM counts than tumors with wild-type EGFR (90% vs. 38.5%). Hence, TAM counts may predict the response to EGFR-TKIs, as TAMs contribute to drug resistance induced by the activity of stromal fibroblasts, as previously demonstrated by Wang et al. in vitro and in vivo [44]. According to the data obtained by two studies that evaluated patients with early and advanced NSCLC, host immunosurveillance is unimpaired in the early stages of NSCLC, when the M1 macrophage phenotype prevails, but falls apart in the advanced stages due to M2 phenotype polarization [45]. Targeted therapy blocks specific signaling pathways, whereas immunotherapy stimulates the immune system to attack tumor cells that previously evaded immune surveillance [46]. Subgroup analysis of clinical trials showed no survival benefit from immunotherapy in patients carrying EGFR mutations [47][48]. EGFR activation increases PD-L1 expression in tumor cells, inducing T-cell apoptosis and immune escape [46]. EGFR-TKIs strengthen MHC class I and II antigen presentation in response to IFN-γ, increasing T-cell-mediated tumor killing [49][50]. These findings explain the potential synergistic effects of immunotherapy and TKIs. However, the clinical benefits of such a combination may be limited [46]. Non-inflamed tumors lack significant lymphocyte infiltration, exhibit low PD-L1 expression, and have elevated levels of immunosuppressive elements in the TME, so such tumors may not be particularly sensitive to immunotherapy [51][52]. EGFR-TKIs modify the TME, weakening the suppressive activity of Tregs and promoting activity of cytotoxic T-cells [46]. EGFR-TKIs were demonstrated to boost the levels of cytotoxic CD8+ T cells and DCs, eliminate FOXP3+ Tregs, and inhibit macrophage polarization to the M2 phenotype, albeit only for a short time. However, EGFR-TKIs also decrease PD-L1 expression in cancer cells. Therefore, a combination of TKIs and immunotherapy may have suboptimal synergistic effects. Tariq et al. observed that inhibition of the STAT6 pathway by gefitinib prevented M2 polarization, but no dynamic changes during TKI treatment were evaluated [53]. Nevertheless, after a long period of TKI treatment, IL-10 activated the STAT3 pathway in MDSCs, inducing Treg activity, inhibiting DCs, and increased M2 macrophage polarization. Thus, the initial effect of gefitinib was neutralized [46].
References
- Macciò, A.; Madeddu, C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology 2020, 159, 357–364.
- Mascaux, C.; Angelova, M.; Vasaturo, A.; Beane, J.; Hijazi, K.; Anthoine, G.; Buttard, B.; Rothe, F.; Willard-Gallo, K.; Haller, A.; et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 2019, 571, 570–575.
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061.
- Williams, L.M.; Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007, 8, 277–284.
- Di Pilato, M.; Kim, E.Y.; Cadilha, B.; Prüßmann, J.N.; Nasrallah, M.N.; Seruggia, D.; Usmani, S.; Misale, S.; Zappulli, V.; Carrizosa, E.; et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 2019, 570, 112–116.
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558.
- Gallimore, A.; Quezada, S.A.; Roychoudhuri, R. Regulatory T cells in cancer: Where are we now? Immunology 2019, 157, 187–189.
- Ayala, M.A.M.; Li, Z.; DuPage, M. Treg programming and therapeutic reprogramming in cancer. Immunology 2019, 157, 198–209.
- Yang, X.O.; Nurieva, R.; Martinez, G.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity 2008, 29, 44–56.
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274.
- Wan, Y.Y.; Flavell, R.A. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007, 445, 766–770.
- Skuljec, J.; Jirmo, A.C.; Habener, A.; Talbot, S.R.; Pul, R.; Grychtol, R.; Aydin, M.; Kleinschnitz, C.; Happle, C.; Hansen, G. Absence of Regulatory T Cells Causes Phenotypic and Functional Switch in Murine Peritoneal Macrophages. Front. Immunol. 2018, 9, 2458.
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26.
- Harris, R.A. Spatial, Temporal, and Functional Aspects of Macrophages during “The Good, the Bad, and the Ugly” Phases of Inflammation. Front. Immunol. 2014, 5, 612.
- Italiani, P.; Boraschi, D.; Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514.
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247.
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499.
- Madeddu, C.; Gramignano, G.; Kotsonis, P.; Coghe, F.; Atzeni, V.; Scartozzi, M.; Maccio, A. Microenvironmental M1 tumor-associated macrophage polarization influences cancer-related anemia in advanced ovarian cancer: Key role of interleukin-6. Haematologica 2018, 103, e388–e391.
- Vargas, A.; Harris, A.J.V.C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Cancer 2016, 16, 525–537.
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469.
- Yeh, H.-H.; Lai, W.-W.; Chen, H.H.W.; Liu, H.-S.; Su, W.-C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309.
- Mondal, A.M.; Horikawa, I.; Pine, S.R.; Fujita, K.; Morgan, K.M.; Vera, E.; Mazur, S.J.; Appella, E.; Vojtesek, B.; Blasco, M.A.; et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Investig. 2013, 123, 5247–5257.
- Maccio, A.; Sanna, E.; Neri, M.; Oppi, S.; Madeddu, C. Cachexia as Evidence of the Mechanisms of Resistance and Tolerance during the Evolution of Cancer Disease. Int. J. Mol. Sci. 2021, 22, 2890.
- Bruno, D.; Dowlati, A. Immunotherapy in EGFR mutant non-small cell lung cancer: When, who and how? Transl. Lung Cancer Res. 2019, 8, 710–714.
- Mansuet-Lupo, A.; Alifano, M.; Pécuchet, N.; Biton, J.; Becht, E.; Goc, J.; Germain, C.; Ouakrim, H.; Régnard, J.F.; Cremer, I.; et al. Intratumoral immune cell densities are associated with lung adenocarcinoma gene alterations. Am. J. Respir. Crit. Care Med. 2016, 194, 1403–1412.
- Lee, C.K.; Man, J.; Lord, S.J.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.-H. Checkpoint Inhibitors in Metastatic EGFR- Mutated Non–Small Cell Lung Cancer—A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407.
- Tuminello, S.; Veluswamy, R.; Lieberman-Cribbin, W.; Gnjatic, S.; Petralia, F.; Wang, P.; Flores, R.; Taioli, E. Prognostic value of immune cells in the tumor microenvironment of early-stage lung cancer: A meta-analysis. Oncotarget 2019, 10, 7142–7155.
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937.
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461.
- Lisberg, A.; Cummings, A.; Goldman, J.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145.
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.-C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492.
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539.
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050.
- Watanabe, H.; Ohashi, K.; Nishii, K.; Seike, K.; Makimoto, G.; Hotta, K.; Maeda, Y.; Kiura, K. A Long-term Response to Nivolumab in a Case of PD-L1-negative Lung Adenocarcinoma with an EGFR Mutation and Surrounding PD-L1-positive Tumor-associated Macrophages. Intern. Med. 2019, 58, 3033–3037.
- Teng, F.; Meng, X.; Kong, L.; Yu, J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: A systematic review. Cancer Lett. 2018, 414, 166–173.
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846.
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567.
- Matsumoto, Y.; Sawa, K.; Fukui, M.; Oyanagi, J.; Izumi, M.; Ogawa, K.; Suzumura, T.; Watanabe, T.; Kaneda, H.; Mitsuoka, S.; et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with EGFR-mutant non-small cell lung cancer. Cancer Sci. 2019, 110, 3244–3254.
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145.
- Su, S.; Dong, Z.-Y.; Xie, Z.; Yan, L.-X.; Li, Y.-F.; Su, J.; Liu, S.-Y.; Yin, K.; Chen, R.-L.; Huang, S.-M.; et al. Strong Programmed Death Ligand 1 Expression Predicts Poor Response and De Novo Resistance to EGFR Tyrosine Kinase Inhibitors Among NSCLC Patients With EGFR Mutation. J. Thorac. Oncol. 2018, 13, 1668–1675.
- Yoneshima, Y.; Ijichi, K.; Anai, S.; Ota, K.; Otsubo, K.; Iwama, E.; Tanaka, K.; Oda, Y.; Nakanishi, Y.; Okamoto, I. PD-L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer 2018, 118, 36–40.
- Takashima, Y.; Sakakibara-Konishi, J.; Hatanaka, Y.; Hatanaka, K.C.; Ohhara, Y.; Oizumi, S.; Hida, Y.; Kaga, K.; Kinoshita, I.; Dosaka-Akita, H.; et al. Clinicopathologic Features and Immune Microenvironment of Non–Small-cell Lung Cancer With Primary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Lung Cancer 2018, 19, 352–359.e1.
- Biswas, S.K.; Lewis, C.E. NF-κB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 2010, 88, 877–884.
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to Stromal Fibroblasts Induces Resistance of Lung Cancer to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638.
- Chung, F.-T.; Lee, K.-Y.; Wang, C.-W.; Heh, C.-C.; Chan, Y.-F.; Chen, H.-W.; Kuo, C.-H.; Feng, P.-H.; Lin, T.-Y.; Wang, C.-H.; et al. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int. J. Cancer 2012, 131, E227–E235.
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444.
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639.
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Perez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550.
- Garrido, G.; Rabasa, A.; Garrido, C.; Chao, L.; Garrido, F.; Lora, A.M.G.; Sánchez-Ramírez, B. Upregulation of HLA Class I Expression on Tumor Cells by the Anti-EGFR Antibody Nimotuzumab. Front. Pharmacol. 2017, 8, 595.
- Pollack, B.P.; Sapkota, B.; Cartee, T.V. Epidermal Growth Factor Receptor Inhibition Augments the Expression of MHC Class I and II Genes. Clin. Cancer Res. 2011, 17, 4400–4413.
- Dong, Z.-Y.; Zhang, J.-T.; Liu, S.-Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.-Y.; Xu, C.-R.; Yan, L.-X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Onco. Immunol. 2017, 6, e1356145.
- Li, H.Y.; McSharry, M.; Bullock, B.; Nguyen, T.T.; Kwak, J.; Poczobutt, J.M.; Sippel, T.R.; Heasley, L.E.; Weiser-Evans, M.C.; Clambey, E.T.; et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol. Res. 2017, 5, 767–777.
- Tariq, M.; Zhang, J.-Q.; Liang, G.-K.; He, Q.-J.; Ding, L.; Yang, B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol. Sin. 2017, 38, 1501–1511.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
05 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No