 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elena Crisa | + 5586 word(s) | 5586 | 2020-09-22 11:02:32 | | | |
| 2 | Vicky Zhou | -2731 word(s) | 2855 | 2020-10-09 07:57:05 | | | | |
| 3 | Vicky Zhou | Meta information modification | 2855 | 2020-10-09 08:35:08 | | | | |
| 4 | Vicky Zhou | Meta information modification | 2855 | 2020-10-12 02:43:11 | | | | |
| 5 | Vicky Zhou | Meta information modification | 2855 | 2020-10-27 05:12:30 | | |
Video Upload Options
Atypical chronic myeloid leukemia, BCR-ABL1 negative (aCML) is a rare myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) with a high rate of transformation to acute myeloid leukemia, and poor survival. Until now, the diagnosis has been based on morphological grounds only, possibly making the real frequency of the disease underestimated. Only recently, new insights in the molecular biology of MDS/MPN syndromes have deepened our knowledge of aCML, enabling us to have a better molecular profile of the disease. The knowledge gleaned from next generation sequencing has complemented morphologic and laboratory WHO criteria for myeloid neoplasms and can provide greater specificity in distinguishing aCML from alternative MDS/MPN or MPNs. The most commonly mutated genes (> 20%) in aCML are SETBP1, ASXL1, N/K-RAS, SRSF2, and TET2, and less frequently (< 10%) CBL, CSFR3, JAK2, EZH2, and ETNK1. Several of these mutations affect the JAK-STAT, MAPK, and ROCK signaling pathways, which are targetable by inhibitors that are already in clinical use and may lead to a personalized treatment of aCML patients unfit for allogeneic transplant, which is currently the only curative option for fit patients.
1. Introduction
Atypical chronic myeloid leukemia (aCML) is a rare BCR-ABL1 negative myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) with a high rate of transformation to acute myeloid leukemia (AML), and is historically characterized by poor survival. The challenges of aCML management affect both diagnosis, due to the heterogeneity of the disease clinical features and the absence of unique biomarkers, and treatment choices, since no current standards of care exist. Median age at presentation is around 70 year old[1]. and median overall survival (OS) is between 12 and 25 months . Almost 40% of patients progress to secondary acute myeloid leukemia (sAML), with a median time to leukemic evolution of 11.2 months[2].
2. Diagnosis and Treatment
2.1 Diagnosis
Atypical CML is a challenging myeloid malignancy with features of both myeloproliferative and myelodysplastic syndromes. The MDS/MPN category was introduced in the 2011 WHO classification to include myeloid neoplasms with clinical, laboratory, and morphologic features that overlap MDS and MPN[3][4]. In addition to aCML, this subgroup includes chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), and a provisional entity within the MDS/MPN unclassifiable group, termed as refractory anemia with ring sideroblasts and thrombocytosis (RARS-T)[4]
The main feature characterizing aCML is the presence of neutrophilic leukocytosis and marked dysgranulopoiesis. Moreover, to fulfil the diagnostic criteria, WBC should be ≥13 × 109/L with ≥10% of immature granulocytes and ≤20% blasts in the blood and the BM. Basophilia and monocytosis may be present, but, unlike Ph+ CML, basophils are minimally increased, and, unlike CMML, monocytes are less than 10% of the leukocytes. To rule out other myeloproliferative disorders, BCR-ABL1 rearrangement should be excluded in all cases, whereas PDGFRA, PDGFRB, FGFR1 rearrangements or PCM1-JAK2 fusions must be excluded if eosinophilia is present[4] (Table 1).
Table 1. WHO 2016 diagnostic criteria for aCML[3].
|
WHO 2016 diagnostic criteria for aCML |
|
• Peripheral blood leukocytosis (WBC count ≥13 × 109/L) because of increased numbers of neutrophils and their precursors with prominent dysgranulopoiesis |
|
• Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥10% of leukocytes |
|
• No Ph chromosome or BCR-ABL1 fusion gene and not meeting criteria for PV, ET, or PMF* |
|
• No evidence of PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1-JAK2 |
|
• Minimal absolute basophilia; basophils usually <2% of leukocytes |
|
• No or minimal absolute monocytosis; monocytes usually <10% of leukocytes |
|
• Hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages |
|
• Less than 20% blasts in the blood and bone marrow |
|
ET, essential thrombocythemia; Ph, Philadelphia; PMF, primary myelofibrosis; PV, polycythemia vera; WBC, white blood cell. |
|
*Cases of myeloproliferative neoplasms (MPN), particularly those in accelerated phase and/or in postpolycythemic or postessential thrombocythemic myelofibrosis, if neutrophilic, may simulate aCML. A previous history of MPN, the presence of MPN features in the bone marrow and/or MPN-associated mutations (in JAK2, CALR, or MPL) tend to exclude a diagnosis of aCML. Conversely, a diagnosis of aCML is supported by the presence of SETBP1 and/or ETNK1 mutations. The presence of a CSF3R mutation is uncommon in aCML and, if detected, should prompt a careful morphologic review to exclude an alternative diagnosis of chronic neutrophilic leukemia or other myeloid neoplasm.1 |
The diagnostic work up should start with complete blood count with manual differential, morphological examination of PB smear, BM examination with assessment of dysplastic features, cytogenetic analysis and FISH to exclude Ph chromosome and rearrangements involving PDGFRA (4q12), PDGFRB (5q31-33), FGFR1 (8p11) or JAK2 (9p24). Once CML has been excluded, second level testing should include human leukocyte antigen (HLA) typing in transplant eligible patients, and assessment with a NGS myeloid panel, not only to confirm diagnosis but also to open the possibility of a target therapy.
2.1.1. Cytogenetics in aCML
The molecular features of aCML include an increased frequency of gene fusions or an aneuploid karyotype. One or two chromosomal aberrations, e.g. trisomy 8 or 9, del (20q), -7 / 7q or isochromosomes 17q, are found in up to 50% of patients[1][5] but are non-specific and similar to cytogenetic findings detectable also in MDS.
2.1.2. Molecular landscape of aCML
The mutational landscape of MDS/MPN represents an overlap between those of MDS and MPN. Similar to MDS, the majority of cases of MDS/MPN harbors at least two mutations involving epigenetic regulation and splicing factors in the dominant clone, with acquisition of additional mutations over time [6].The most commonly affected genes (> 20%) in aCML are SETBP1, ASXL1, N/K-RAS, SRSF2, and TET2, and less frequently (< 10%) CBL, CSFR3, JAK2, EZH2 and ETNK1[7][1][8][9][10][11][12][13][14](Figure 1). Usually the JAK2, CALR and MPL genes are wild type, although a few cases of CMML and aCML have been reported to harbor JAK2 mutations[15][16] .
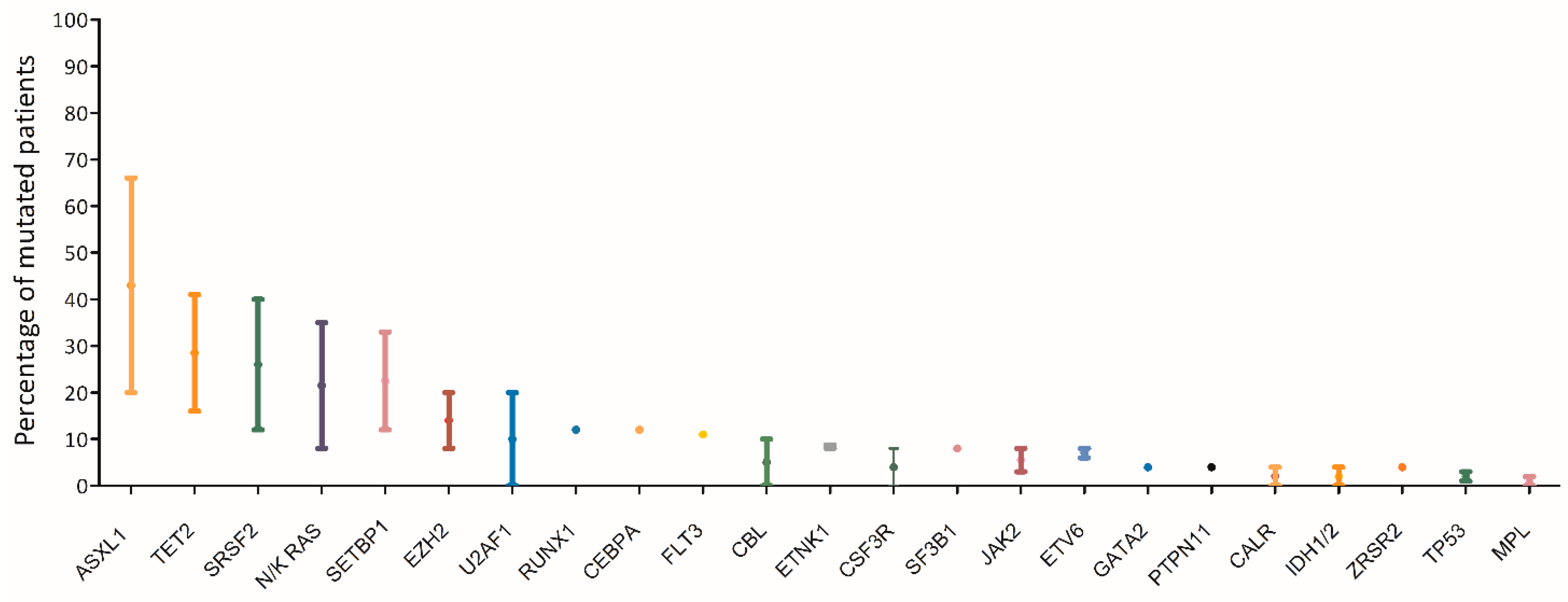
Figure 1. Mutational landscape of aCML. The main genes found to be mutated in aCML are represented on the x axis of the graph. Bars represent the minimum and maximum percentage of mutated patients for each gene reported in different study populations[7][1][8][9][10][11][12][13][14].
Mutations in SETBP1 and ETNK1 appear to be the alterations most closely associated with aCML, even though they are not univocally disease-specific[4][5][11][12][17]. As highlighted by the WHO-2016 revised criteria, mutations in SETBP1 and ETNK1 may be useful diagnostic tools for the disease. By contrast, mutations in the CSF3R gene, initially described in both CNL and aCML[18], appear to be more specifically associated with CNL, in which these mutations occur in 50 to 80% of cases according to different patient series.
In the last decade, the identification of oncogenic mutations in signal transduction proteins has clarified that specific pathways regulating the proliferation of myeloid lineage cells may be deregulated in leukemia [19]. Several mutations commonly found in aCML impact on the JAK-STAT, MAPK and ROCK signaling pathways, that are known to be responsible for myeloproliferation in physiological conditions and to be aberrantly activated in myeloproliferative diseases [20], including aCML[21][19] (Figure 2).
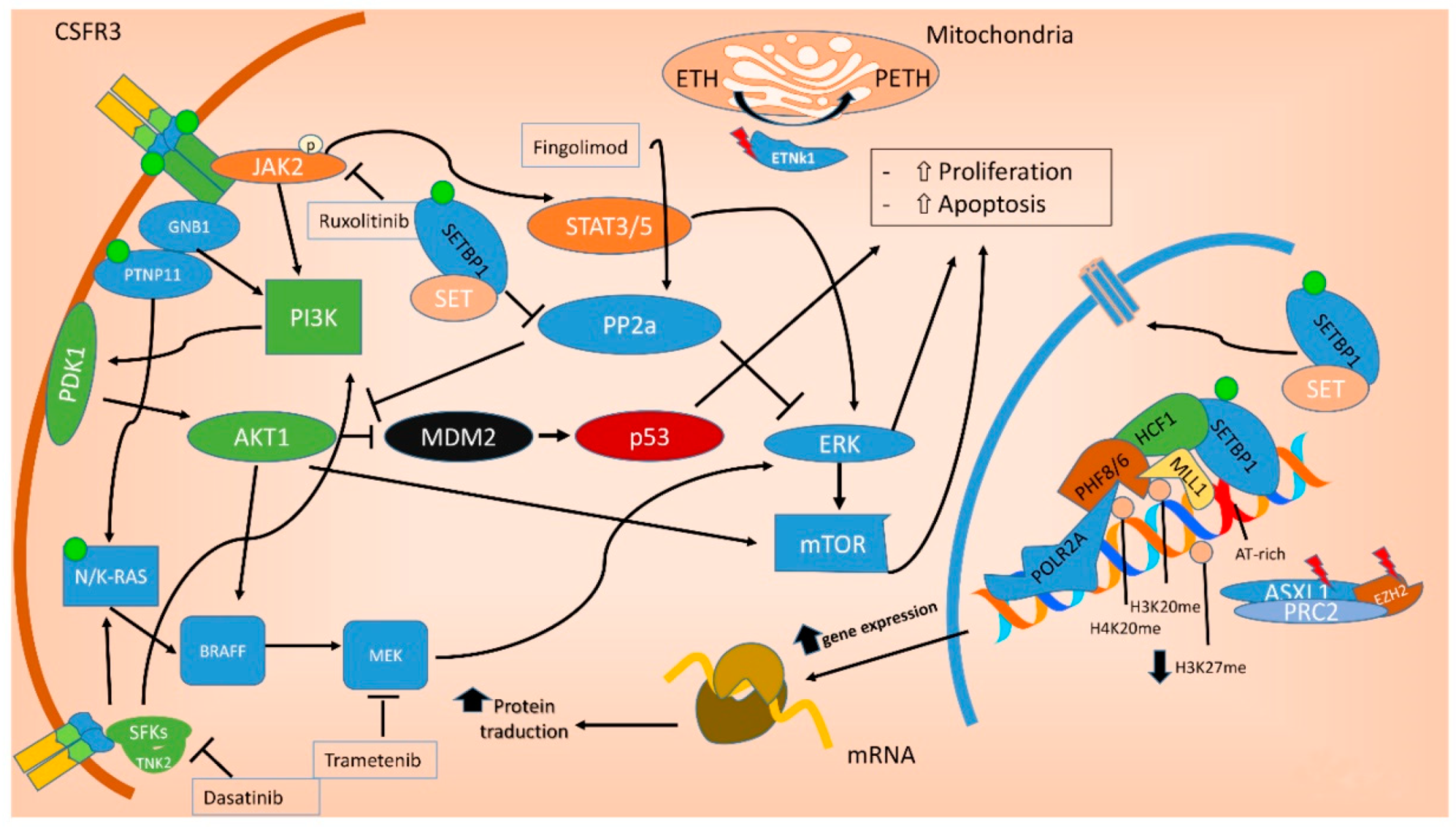
Figure 2. Molecular pathways involved in aCML. Mutations in the ASXL1 and EZH2 genes are mainly point mutations that lead to impaired function of the polycomb repressive complex 2 (PRC2), which translate in decreased epigenetic repression of key genes involved in stem cell renewal, thus promoting myeloid proliferation and differentiation. Mutations of both NRAS and PTNP11 result in a constitutive activation of MAPK, promoting cancer cell survival and proliferation. SETBP1 encodes a protein named SET binding protein 1 (SEB) that regulates the SET inhibitory activity on tumor suppressors, including PP2A. In aCML, all the SETBP1 mutations result in an increased gene expression and, through SET, in a reduction of PP2A inhibitory activity on AKT and MAPK pathways, leading to increased cellular proliferation and survival. Fingolimod targets PP2A with an activating effect. ETNK1 encodes an ethanolamine kinase, which catalyzes the first step of the de novo phosphatidylethanolamine biosynthesis pathway, critical for regulating membrane architecture and the topology of transmembrane domains of membrane binding proteins. Due to the fact that the ethanolamine kinase 1 contributes to different processes in the cell, the mechanisms by which the mutant protein induces myeloproliferation have not yet been clarified. CSFR3 mutations may be membrane proximal mutations or truncation mutations or a combination of the two. All the activating missense mutations target the proximal domain leading to increased dimerization and activation of JAK-STAT pathway, sensitive to its kinase inhibitor ruxolitinib. Conversely, CSFR3 truncating mutations induce receptor signaling through SRC family kinase rendering the cells sensitive to the multikinase inhibitor dasatinib. Green dot: activating mutation; red lightning: inactivating mutations. ETH: Ethanolamine; PETH: phosphatidylethanolamine.
2.2. Treatment
No standard of care is currently available for aCML management. This rare entity of MDS/MPN, as already detailed, is characterized by heterogeneous clinical and genetic features with a very poor prognosis. In addition, due to the low disease incidence and the absence of large randomized clinical trials, the physician’s choices remain challenging. Several treatment strategies validated in other myeloid diseases have been evaluated in aCML, starting from erythropoiesis stimulating agents (ESAs) to improve the anemia and up to cytoreductive drugs to control the proliferation and reduce the blast count (AML-like chemotherapy, hypomethylating agents (HMA), hydroxyurea (HU), or PEG–IFN–α. Given the recent clarification of the mutational landscape of MDS/MPN, different target therapies, including JAK2 inhibitors, are currently being investigated. However, up to date, the only curative treatment remains HSCT, an option available for the younger patients only. Participation on clinical trials should be considered in all cases. A possible treatment algorithm is reported in Figure 3.
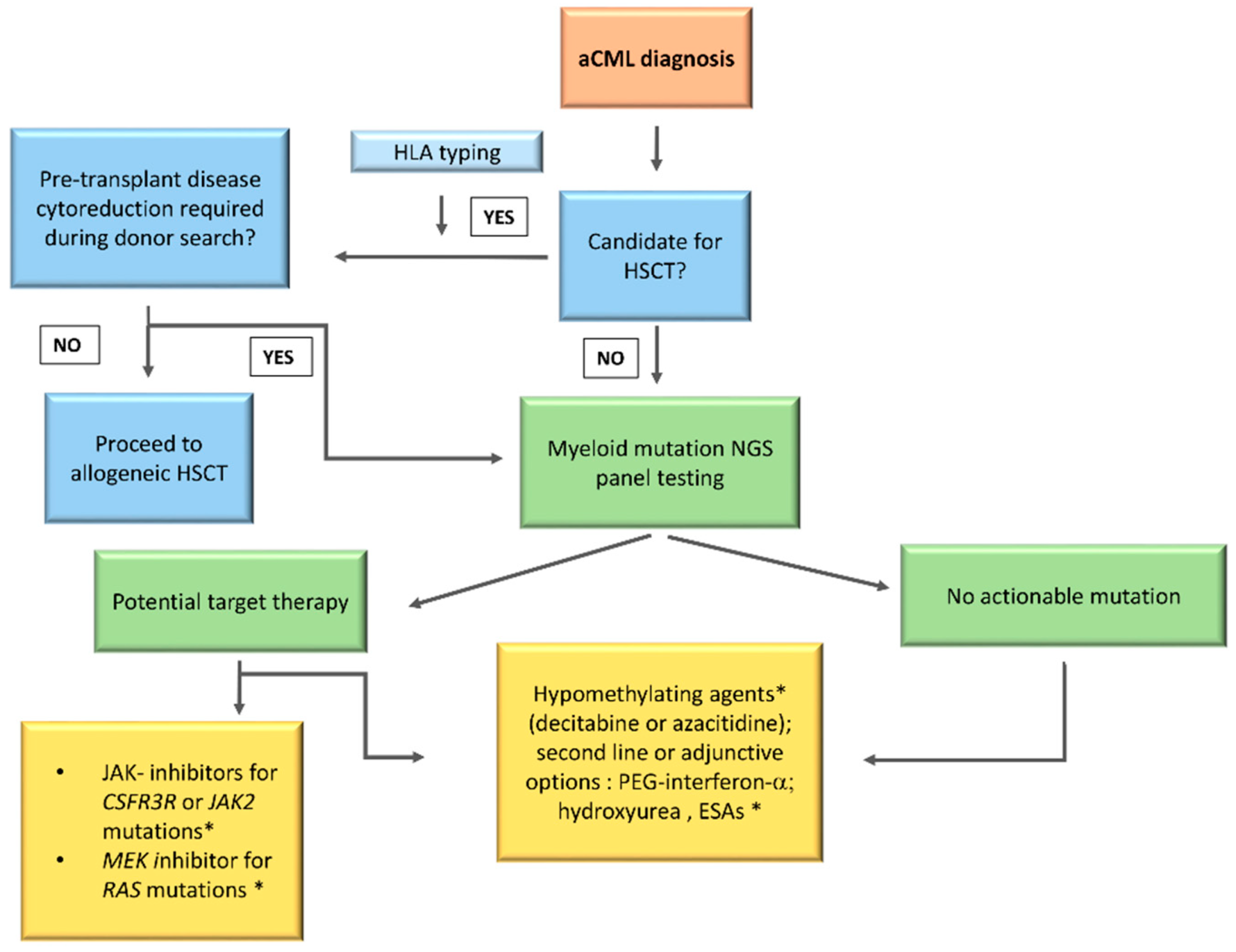
2.2.1. Hematopoietic Stem Cell Transplant
HSCT is considered as the sole potentially curative strategy for aCML. Data on this approach are scant and most evidence originates from studies including patients with heterogeneous MDS/MPNs. Lim et al. reported a series of 10 patients with MDS/MPN who received HSCT. The 5-year OS and disease-free survival (DFS) rates of the whole cohort were 42.2% and 46%, respectively. Only 2 out of 10 patients were aCML, and, at the median follow-up of 47.5 months, both were alive and in remission [22]. Similar OS and DFS rates (2 years OS and DFS 47% and 37%, respectively) were reported by Mittal et al. in a patient series of 20 MPD/MPN cases, including 8 aCML, who received HSCT [23]. Koldehoff et al. analyzed 9 aCML patients, receiving either sibling allogeneic donor type (n = 4) or MUD (n = 4) or a syngeneic transplantation from a twin sibling (n = 1) [24]. All patients were in complete remission at the median follow-up of 55 months and only one patient relapsed 19 months after syngenic HSCT but re-obtained a remission after a second allotransplant [25]. A subsequent follow-up study including 21 aCML transplanted patients reported a 5 year OS rate of 80% with a median survival of 48 months [26]. These promising results with HSCT were confirmed in a larger series of 42 aCML cases reported by Onida et al. who received HLA-identical sibling (64%) or MUD (36%) cells [27]. At a median follow-up of 89 months, 87% of patients were in complete remission with a 5 year OS rate of 51% and a relapse free survival rate of 36%.
No evidence-based information on the best timing of transplantation is currently available, and indications rely on expert opinions. Gotlib et al. proposed HSCT for all candidate patients at the time of diagnosis [28], whereas the Moffitt group suggested to stratify patients according to the known clinical (age > 65, leukocytosis > 50 × 109/L) and molecular (SETBP1 mutation) prognostic factors and to offer HSCT at diagnosis in high risk cases and defer the procedures in the others [29].
The choice between these two approaches should also consider the availability of a suitable donor at the time of diagnosis and the presence of a targetable mutation that might be appropriate for an attempt with molecular therapies.
In conclusion, allogeneic HSCT may be considered a promising approach for a subset of aCML patients and the new molecular findings may pave the way to a MRD driven approach to treat patients after HSCT; however, a large number of cases are elderly and may be not candidates for the HSCT.
2.2.2. Hypomethylating Agents
Based on the use of the HMA azacytidine or decitabine in CMML, different studies have tried to use these drugs also in aCML [30]. Kantarjan et al. analyzed the role of decitabine in a large series of 130 patients with BCR/ABL1 positive and negative CML. Out of seven aCML patients, four obtained a clinically meaningful response but their median survival was only 13 months and the 2 year survival rate was 14% [31]. More recently, single case reports have documented a response to decitabine in a CML patients, that in 2 cases successfully allowed a bridge to transplant [32][33][34][35]. Data on azacitidine are even more limited and less promising. Patnaik et al. reported the outcome of 4 patients who were treated with azacytidine and achieved a stable disease as best response. No patients underwent HSCT in this small series.
HMA cannot be considered as a standard of care for aCML and their use is off-label. However, these drugs represent an alternative to standard chemotherapy for the pre-transplant cytoreduction in case of a low blast count, as in our clinical case 1, or in the presence of comorbidities. Moreover, given the evidence that the use of HMA as a bridge to HSCT in MDS does not worsen the transplant outcome and OS after transplant, it is tempting to extrapolate this hypothesis also to aCML [36][37][38]. However, it must be emphasized that the response to this agent is transitory, so even if a CR is reached the transplant must not be delayed.
HMA may also be considered for patients who are ineligible to transplant that do not carry actionable somatic mutations and are intolerant to HU as an alternative to IFN.
2.2.3. AML-Like Chemotherapy
The role of AML-like intensive chemotherapy has not been explored extensively, but is usually offered to selected patients with high-risk proliferative clinical behavior as a bridge to HSCT [29].
2.2.4. Interferon-Alpha and Hydroxyurea
HU is commonly used to control splenomegaly and leukocytosis. Older studies reported that HU and IFN-alfa are able to induce a complete hematological response, however with short duration [39][40]. PEG–IFN–α–2b is a pegylated IFN with a significant advantage over non-pegylated form in the administration schedule (once a week) and toxicity profile. In a study on 38 patients with BCR–ABL–negative MPDs, which included 5 aCML, 45% of patients achieved a complete or partial response, with a median duration of response of 20 months [41]. Both HU and IFN are useful in a palliative setting, where an allogenic stem-cell transplant is not considered due to age or comorbidities. In these clinical contexts, HU and IFN may help control myeloproliferation, reduce splenomegaly and possibly, in the case of IFN, control symptoms (as in our case 2). The pegilated form of IFN should be preferred whenever possible for the patient’s convenience.
2.2.5. Target Therapy
Several promising targeted therapies are currently being investigated, including the JAK inhibitor ruxolitinib, the SRC kinase inhibitor dasatinib, and the MEK inhibitor trametinib (the main molecular targetable pathways in aCML are shown in Figure 2).
JAK2 V617F and CSF3R T618I mutated cases, although infrequent among aCML patients, might benefit from the JAK2 inhibitor ruxolitinib, that is already in clinical use for the treatment of myelofibrosis and polycythemia vera. Preclinical studies have shown that membrane proximal mutation of CSF3R, like T618I, induce a lethal MPN similar to aCML and CNL in murine models, and that the administration of ruxolitinib is effective in reducing WBC, decreasing spleen weight and increasing body weight in mice [42][43]. Based on these promising results, ruxolitinib has been tested as a single agent in a few case reports with encouraging results [44][45]. Recently, in a phase II study on a series of 35 MDS/MPN, including 4 aCML, the combination of ruxolitinib and azacitidine was well tolerated and led to 57% of responses, particularly in JAK2 mutated patients. A benefit in survival, more evident in MDS/MPN–U than in aCML, was observed. Of note, no CSF3R mutated patients were included in this study [46]. The results of ruxolitinib in continuum or as a bridge to allotransplant in primary myelofibrosis may further suggest its use in aCML [47][48], however, due to the limited data currently available for aCML, this agent should be recommended in the setting of clinical trials only.
3. Conclusions
The diagnosis of aCML is always a difficult task to accomplish. Until now, the diagnosis has been based on morphological grounds only, possibly underestimating the true frequency of the disease. Only recently, new insights in the molecular biology of MDS/MPN syndromes have deepened our knowledge in aCML. However, all the mutations described in aCML so far are neither specific nor diagnostic, being present in a wide range of other hematological malignancies and also in the normal elderly population, even though at a very low VAF. With a more comprehensive molecular profiling, the diagnostic criteria will be hopefully refined in the future to include also genetic features resulting in decreased reliance on absolute cutoffs in blood counts that may vary along with the disease history of the patients. Nonetheless, as discussed earlier, in some cases these alterations can be more than a simple diagnostic test and may allow personalized treatment. Large collaborative efforts are needed to design studies that will provide data so that the management of these diseases will be evidence-driven.
References
- Patnaik, M.M.; Barraco, D.; Lasho, T.L.; Finke, C.M.; Reichard, K.; Hoversten, K.P.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Targeted next generation sequencing and identification of risk factors in World Health Organization defined atypical chronic myeloid leukemia. Am. J. Hematol. 2017, 92, 542–548.
- M. Breccia, F. Biondo, R. Latagliata, I. Carmosino, F. Mandelli, G. Alimena, Identification of risk factors in atypical chronic myeloid leukemia., Haematologica. 91 (2006) 1566–8. http://www.ncbi.nlm.nih.gov/pubmed/17043019 (accessed June 4, 2020).
- J.W. Vardiman, J. Thiele, D.A. Arber, R.D. Brunning, M.J. Borowitz, A. Porwit, N.L. Harris, M.M. Le Beau, E. Hellström-Lindberg, A. Tefferi, C.D. Bloomfield, The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes., Blood. 114 (2009) 937–51. https://doi.org/10.1182/blood-2009-03-209262.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- M. Meggendorfer, U. Bacher, T. Alpermann, C. Haferlach, W. Kern, C. Gambacorti-Passerini, T. Haferlach, S. Schnittger, SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations., Leukemia. 27 (2013) 1852–60. https://doi.org/10.1038/leu.2013.133.
- H. Zhang, B. Wilmot, D. Bottomly, K.-H.T. Dao, E. Stevens, C.A. Eide, V. Khanna, A. Rofelty, S. Savage, A. Reister Schultz, N. Long, L. White, A. Carlos, R. Henson, C. Lin, R. Searles, R.H. Collins, D.J. DeAngelo, M.W. Deininger, T. Dunn, T. Hein, M.R. Luskin, B.C. Medeiros, S.T. Oh, D.A. Pollyea, D.P. Steensma, R.M. Stone, B.J. Druker, S.K. McWeeney, J.E. Maxson, J.R. Gotlib, J.W. Tyner, Genomic landscape of neutrophilic leukemias of ambiguous diagnosis, Blood. 134 (2019) 867–879. https://doi.org/10.1182/blood.2019000611.
- S.A. Wang, R.P. Hasserjian, P.S. Fox, H.J. Rogers, J.T. Geyer, D. Chabot-Richards, E. Weinzierl, J. Hatem, J. Jaso, R. Kanagal-Shamanna, F.C. Stingo, K.P. Patel, M. Mehrotra, C. Bueso-Ramos, K.H. Young, C.D. Dinardo, S. Verstovsek, R. V. Tiu, A. Bagg, E.D. Hsi, D.A. Arber, K. Foucar, R. Luthra, A. Orazi, Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms, Blood. 123 (2014) 2645–2651. https://doi.org/10.1182/blood-2014-02-553800.
- J. Gotlib, How I treat atypical chronic myeloid leukemia, Blood. 129 (2017) 838–845. https://doi.org/10.1182/blood-2016-08-693630.
- T. Lasho, Atypical CML- the role of morphology and precision genomics, Best Pract. Res. Clin. Haematol. 33 (2020) 101133. https://doi.org/https://doi.org/10.1016/j.beha.2019.101133.
- C.B. Gambacorti-Passerini, C. Donadoni, A. Parmiani, A. Pirola, S. Redaelli, G. Signore, V. Piazza, L. Malcovati, D. Fontana, R. Spinelli, V. Magistroni, G. Gaipa, M. Peronaci, A. Morotti, C. Panuzzo, G. Saglio, E. Usala, D.-W. Kim, D. Rea, K. Zervakis, N. Viniou, A. Symeonidis, H. Becker, J. Boultwood, L. Campiotti, M. Carrabba, E. Elli, G.R. Bignell, E. Papaemmanuil, P.J. Campbell, M. Cazzola, R. Piazza, Recurrent ETNK1 mutations in atypical chronic myeloid leukemia, Blood. 125 (2015) 499–503. https://doi.org/10.1182/blood-2014-06-579466.
- M. Meggendorfer, T. Haferlach, T. Alpermann, S. Jeromin, C. Haferlach, W. Kern, S. Schnittger, Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia, Haematologica. 99 (2014) e244–e246. https://doi.org/10.3324/HAEMATOL.2014.113159.
- R. Piazza, S. Valletta, N. Winkelmann, S. Redaelli, R. Spinelli, A. Pirola, L. Antolini, L. Mologni, C. Donadoni, E. Papaemmanuil, S. Schnittger, D.W. Kim, J. Boultwood, F. Rossi, G. Gaipa, G.P. De Martini, P.F. Di Celle, H.G. Jang, V. Fantin, G.R. Bignell, V. Magistroni, T. Haferlach, E.M. Pogliani, P.J. Campbell, A.J. Chase, W.J. Tapper, N.C.P. Cross, C. Gambacorti-Passerini, Recurrent SETBP1 mutations in atypical chronic myeloid leukemia, Nat. Genet. 45 (2013) 18–24. https://doi.org/10.1038/ng.2495.
- F. Onida, G. Ball, H.M. Kantarjian, T.L. Smith, A. Glassman, M. Albitar, B. Scappini, M.B. Rios, M.J. Keating, M. Beran, Characteristics and outcome of patients with Philadelphia chromosome negative, bcr/abl negative chronic myelogenous leukemia., Cancer. 95 (2002) 1673–84. https://doi.org/10.1002/cncr.10832.
- T. Klampfl, H. Gisslinger, A.S. Harutyunyan, H. Nivarthi, E. Rumi, J.D. Milosevic, N.C.C. Them, T. Berg, B. Gisslinger, D. Pietra, D. Chen, G.I. Vladimer, K. Bagienski, C. Milanesi, I.C. Casetti, E. Sant’Antonio, V. Ferretti, C. Elena, F. Schischlik, C. Cleary, M. Six, M. Schalling, A. Schönegger, C. Bock, L. Malcovati, C. Pascutto, G. Superti-Furga, M. Cazzola, R. Kralovics, Somatic mutations of calreticulin in myeloproliferative neoplasms., N. Engl. J. Med. 369 (2013) 2379–90. https://doi.org/10.1056/NEJMoa1311347.
- D.P. Steensma, G.W. Dewald, T.L. Lasho, H.L. Powell, R.F. McClure, R.L. Levine, D.G. Gilliland, A. Tefferi, The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes, Blood. 106 (2005) 1207–1209. https://doi.org/10.1182/blood-2005-03-1183.
- A. V. Jones, S. Kreil, K. Zoi, K. Waghorn, C. Curtis, L. Zhang, J. Score, R. Seear, A.J. Chase, F.H. Grand, H. White, C. Zoi, D. Loukopoulos, E. Terpos, E.-C. Vervessou, B. Schultheis, M. Emig, T. Ernst, E. Lengfelder, R. Hehlmann, A. Hochhaus, D. Oscier, R.T. Silver, A. Reiter, N.C.P. Cross, Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders, Blood. 106 (2005) 2162–2168. https://doi.org/10.1182/blood-2005-03-1320.
- F. Damm, R. Itzykson, O. Kosmider, N. Droin, A. Renneville, V. Chesnais, V. Gelsi-Boyer, S. de Botton, N. Vey, C. Preudhomme, A. Clavert, E. Delabesse, S. Park, D. Birnbaum, M. Fontenay, O.A. Bernard, E. Solary, SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias, Leukemia. 27 (2013) 1401–1403. https://doi.org/10.1038/leu.2013.35.
- J.E. Maxson, J. Gotlib, D.A. Pollyea, A.G. Fleischman, A. Agarwal, C.A. Eide, D. Bottomly, B. Wilmot, S.K. McWeeney, C.E. Tognon, J.B. Pond, R.H. Collins, B. Goueli, S.T. Oh, M.W. Deininger, B.H. Chang, M.M. Loriaux, B.J. Druker, J.W. Tyner, Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML, N. Engl. J. Med. 368 (2013) 1781–1790. https://doi.org/10.1056/NEJMoa1214514.
- K. Zoi, N.C.P. Cross, Molecular pathogenesis of atypical CML, CMML and MDS/MPN-unclassifiable, Int. J. Hematol. 101 (2015) 229–242. https://doi.org/10.1007/s12185-014-1670-3.
- S. Rocca, G. Carrà, P. Poggio, A. Morotti, M. Brancaccio, Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia., Mol. Cancer. 17 (2018) 40. https://doi.org/10.1186/s12943-018-0774-4.
- J. Gotlib, J.E. Maxson, T.I. George, J.W. Tyner, The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment, Blood. 122 (2013) 1707–1711. https://doi.org/10.1182/blood-2013-05-500959.
- Lim, S.N.; Lee, J.H.; Lee, J.H.; Kim, D.Y.; Kim, S.D.; Kang, Y.A.; Lee, Y.S.; Lee, K.H. Allogeneic hematopoietic cell transplantation in adult patients with myelodysplastic/myeloproliferative neoplasms. Blood Res. 2013, 48, 178–184.
- Mittal, P.; Saliba, R.M.; Giralt, S.A.; Shahjahan, M.; Cohen, A.I.; Karandish, S.; Onida, F.; Beran, M.; Champlin, R.E.; de Lima, M. Allogeneic transplantation: A therapeutic option for myelofibrosis, chronic myelomonocytic leukemia and Philadelphia-negative/BCR-ABL-negative chronic myelogenous leukemia. Bone Marrow Transplant. 2004, 33, 1005–1009.
- Koldehoff, M.; Beelen, D.W.; Trenschel, R.; Steckel, N.K.; Peceny, R.; Ditschkowski, M.; Ottinger, H.; Elmaagacli, A.H. Outcome of hematopoietic stem cell transplantation in patients with atypical chronic myeloid leukemia. Bone Marrow Transplant. 2004, 34, 1047–1050.
- H. Hausmann, V.R. Bhatt, J. Yuan, L.J. Maness, A.K. Ganti, Activity of single-agent decitabine in atypical chronic myeloid leukemia, J. Oncol. Pharm. Pract. 22 (2016) 790–794. https://doi.org/10.1177/1078155215605662.
- Koldehoff, M.; Steckel, N.K.; Hegerfeldt, Y.; Ditschkowski, M.; Beelen, D.W.; Elmaagacli, A.H. Clinical course and molecular features in 21 patients with atypical chronic myeloid leukemia. Int. J. Lab. Hematol. 2012, 34, e3–e5.
- Onida, F.; de Wreede, L.C.; van Biezen, A.; Eikema, D.-J.; Byrne, J.L.; Iori, A.P.; Schots, R.; Jungova, A.; Schetelig, J.; Finke, J.; et al. Allogeneic stem cell transplantation in patients with atypical chronic myeloid leukaemia: A retrospective study from the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Br. J. Haematol. 2017, 177, 759–765.
- Gotlib, J. How I treat atypical chronic myeloid leukemia. Blood 2017, 129, 838–845.
- Talati, C.; Padron, E. An Exercise in Extrapolation: Clinical Management of Atypical CML, MDS/MPN-Unclassifiable, and MDS/MPN-RS-T. Curr. Hematol. Malig. Rep. 2016, 11, 425–433.
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2016, 91, 631–642.
- Kantarjian, H.M.; O’Brien, S.; Cortes, J.; Giles, F.J.; Faderl, S.; Issa, J.-P.; Garcia-Manero, G.; Rios, M.B.; Shan, J.; Andreeff, M.; et al. Results of decitabine (5-aza-2′deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer 2003, 98, 522–528.
- Hausmann, H.; Bhatt, V.R.; Yuan, J.; Maness, L.J.; Ganti, A.K. Activity of single-agent decitabine in atypical chronic myeloid leukemia. J. Oncol. Pharm. Pract. 2016, 22, 790–794.
- Mao, L.; You, L.; Yang, M.; Li, Y.; Ye, X.; Tong, H.Y. The First Case of Decitabine Successfully in Treatment of Atypical Chronic Myeloid Leukemia with CEBPA Double Mutation. Chemother. Open Access 2013, 2, 114.
- Tong, X.; Li, J.; Zhou, Z.; Zheng, D.; Liu, J.; Su, C. Efficacy and side-effects of decitabine in treatment of atypical chronic myeloid leukemia. Leuk. Lymphoma 2015, 56, 1911–1913.
- Jiang, H.; Wu, Z.; Ren, L.; Tao, D.; Tong, H. Decitabine for the treatment of atypical chronic myeloid leukemia: A report of two cases. Oncol. Lett. 2016, 11, 689–692.
- Nishihori, T.; Perkins, J.; Mishra, A.; Komrokji, R.; Kim, J.; Kharfan-Dabaja, M.A.; Perez, L.; Lancet, J.; Fernandez, H.; List, A.; et al. Pretransplantation 5-azacitidine in high-risk myelodysplastic syndrome. Biol. Blood Marrow Transplant. 2014, 20, 776–780.
- Gerds, A.T.; Deeg, H.J. Transplantation for myelodysplastic syndrome in the era of hypomethylating agents. Curr. Opin. Hematol. 2012, 19, 71–75.
- Voso, M.T.; Leone, G.; Piciocchi, A.; Fianchi, L.; Santarone, S.; Candoni, A.; Criscuolo, M.; Masciulli, A.; Cerqui, E.; Molteni, A.; et al. Feasibility of allogeneic stem-cell transplantation after azacitidine bridge in higher-risk myelodysplastic syndromes and low blast count acute myeloid leukemia: Results of the BMT-AZA prospective study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1547–1553.
- Kurzrock, R.; Bueso-Ramos, C.E.; Kantarjian, H.; Freireich, E.; Tucker, S.L.; Siciliano, M.; Pilat, S.; Talpaz, M. BCR Rearrangement–Negative Chronic Myelogenous Leukemia Revisited. J. Clin. Oncol. 2001, 19, 2915–2926.
- Martiat, P.; Michaux, J.L.; Rodhain, J. Philadelphia-negative (Ph−) chronic myeloid leukemia (CML): Comparison with Ph+ CML and chronic myelomonocytic leukemia. The Groupe Français de Cytogénétique Hématologique. Blood 1991, 78, 205–211.
- Jabbour, E.; Kantarjian, H.; Cortes, J.; Thomas, D.; Garcia-Manero, G.; Ferrajoli, A.; Faderl, S.; Richie, M.A.; Beran, M.; Giles, F.; et al. PEG-IFN-α-2b therapy in BCR-ABL–negative myeloproliferative disorders. Cancer 2007, 110, 2012–2018.
- Fleischman, A.G.; Maxson, J.E.; Luty, S.B.; Agarwal, A.; Royer, L.R.; Abel, M.L.; MacManiman, J.D.; Loriaux, M.M.; Druker, B.J.; Tyner, J.W. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood 2013, 122, 3628.
- Maxson, J.E.; Luty, S.B.; MacManiman, J.D.; Paik, J.C.; Gotlib, J.; Greenberg, P.; Bahamadi, S.; Savage, S.L.; Abel, M.L.; Eide, C.A.; et al. The Colony Stimulating Factor 3 Receptor T640N mutation is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin. Cancer Res. 2016, 22, 757.
- Dao, K.-H.T.; Solti, M.B.; Maxson, J.E.; Winton, E.F.; Press, R.D.; Druker, B.J.; Tyner, J.W. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk. Res. Reports 2014, 3, 67–69.
- Freedman, J.L.; Desai, A.V.; Bailey, L.C.; Aplenc, R.; Burnworth, B.; Zehentner, B.K.; Teachey, D.T.; Wertheim, G. Atypical Chronic Myeloid Leukemia in Two Pediatric Patients. Pediatr. Blood Cancer 2016, 63, 156–159.
- Assi, R.; Kantarjian, H.M.; Garcia-Manero, G.; Cortes, J.E.; Pemmaraju, N.; Wang, X.; Nogueras-Gonzalez, G.; Jabbour, E.; Bose, P.; Kadia, T.; et al. A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am. J. Hematol. 2018, 93, 277–285.
- Shanavas, M.; Popat, U.; Michaelis, L.C.; Fauble, V.; McLornan, D.; Klisovic, R.; Mascarenhas, J.; Tamari, R.; Arcasoy, M.O.; Davies, J.; et al. Outcomes of Allogeneic Hematopoietic Cell Transplantation in Patients with Myelofibrosis with Prior Exposure to Janus Kinase 1/2 Inhibitors. Biol. Blood Marrow Transplant. 2016, 22, 432–440.
- Shahnaz Syed Abd Kadir, S.; Christopeit, M.; Wulf, G.; Wagner, E.; Bornhauser, M.; Schroeder, T.; Crysandt, M.; Mayer, K.; Jonas, J.; Stelljes, M.; et al. Impact of ruxolitinib pretreatment on outcomes after allogeneic stem cell transplantation in patients with myelofibrosis. Eur. J. Haematol. 2018, 101, 305–317.


