Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | yating Pan | -- | 2020 | 2022-06-29 10:27:53 | | | |
| 2 | Beatrix Zheng | + 3 word(s) | 2023 | 2022-06-29 10:48:31 | | | | |
| 3 | Beatrix Zheng | -24 word(s) | 1999 | 2022-07-01 03:06:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pan, Y.; Cao, W.; Mu, Y.; Zhu, Q. Development of Single-cell RNA Sequencing. Encyclopedia. Available online: https://encyclopedia.pub/entry/24618 (accessed on 08 August 2026).
Pan Y, Cao W, Mu Y, Zhu Q. Development of Single-cell RNA Sequencing. Encyclopedia. Available at: https://encyclopedia.pub/entry/24618. Accessed August 08, 2026.
Pan, Yating, Wenjian Cao, Ying Mu, Qiangyuan Zhu. "Development of Single-cell RNA Sequencing" Encyclopedia, https://encyclopedia.pub/entry/24618 (accessed August 08, 2026).
Pan, Y., Cao, W., Mu, Y., & Zhu, Q. (2022, June 29). Development of Single-cell RNA Sequencing. In Encyclopedia. https://encyclopedia.pub/entry/24618
Pan, Yating, et al. "Development of Single-cell RNA Sequencing." Encyclopedia. Web. 29 June, 2022.
Copy Citation
Single-cell RNA sequencing (scRNA-seq) technology provides a powerful tool for understanding complex biosystems at the single-cell and single-molecule level. The application of droplet- and microwell-based microfluidics in scRNA-seq has contributed greatly to improving sequencing throughput.
scRNA-seq
microfluidics
droplet
microwell
1. General Process of scRNA-seq
Although dozens of different scRNA-seq methods have been published [1][2][3][4], they all follow a similar general process, which is shown in Figure 1. In short, it includes single-cell dissociation and isolation, cell lysis and RNA release, reverse transcription (RT), second-strand cDNA synthesis, amplification, library preparation, sequencing and data analysis. In this section, the researchers will introduce some technical points and solutions in sequencing library construction.
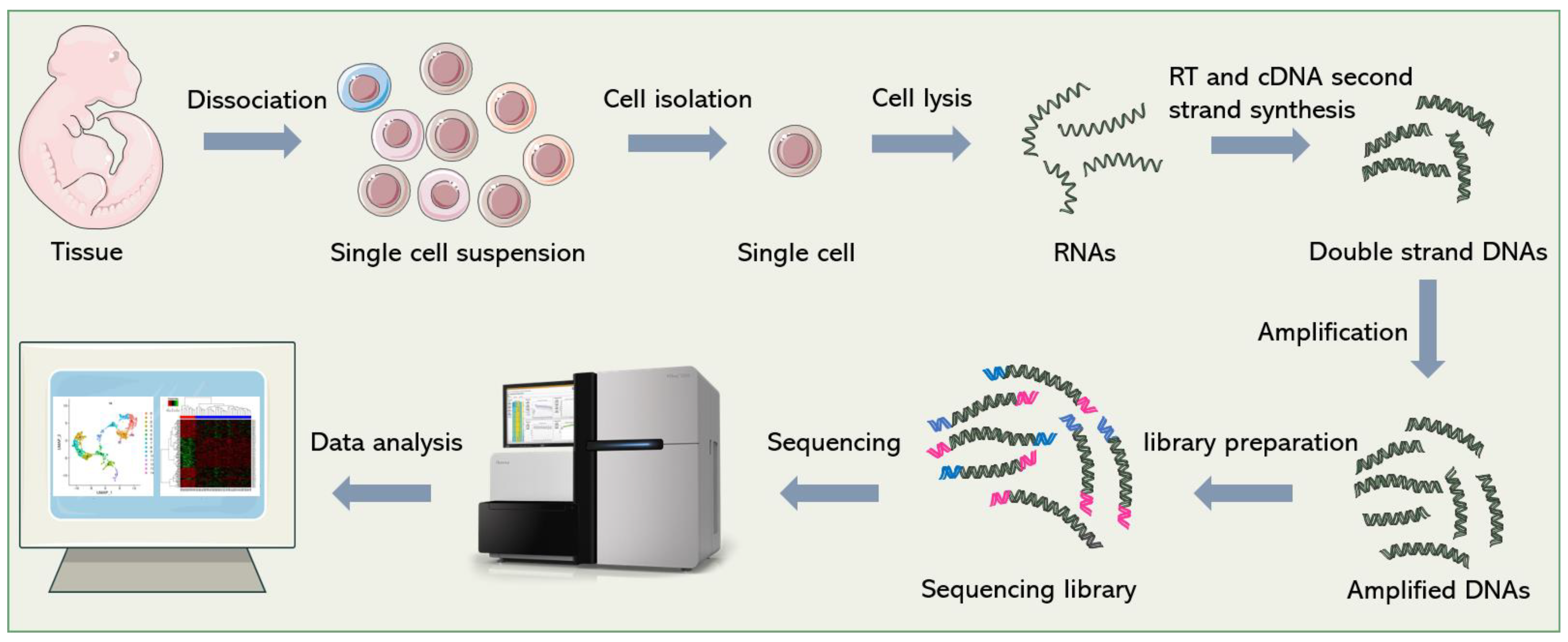
Figure 1. General workflow of scRNA-seq. Firstly, single-cell suspensions are obtained from cultured cells or tissue blocks. Then, cells are isolated and lysed. The released RNAs are reverse-transcribed into cDNAs. After second-strand synthesis and amplification, sequencing adapters are added to both ends of the cDNAs to construct the final sequencing library. Finally, bioinformatics pipelines are used to analyze the sequencing data and re-establish gene expression signatures of single cells.
Specifically, there are two ways of single-cell dissociation: enzymatic and mechanical dissociation [5]. The dosage, digestion time, temperature, and other parameters of enzymatic digestion need to be adjusted according to the property of tissue to maintain the integrity and activity of cells as much as possible. Mechanical dissociation or laser capture microdissection (LCM) uses a microscope and laser beam to carefully dissect single cells from frozen tissue sections [6]. This technology was developed at the National Cancer Institute (NCI), originally for the study of heterogeneous cell populations in tumors [7]. LCM has been widely used for single-cell analysis [8][9][10][11], with the advantage of preserving the spatial information of single cells in tissues and facilitating the analysis of cell–cell and cell–environment interactions [12][13][14]. However, the main restriction that limits the application of LCM in single-cell sequencing is its low throughput. In contrast, a large number of single cells can be easily obtained by enzymatic digestion, which is suitable for high-throughput analysis. However, the spatial information of the cells is inevitably lost in the dissociation step [15]. In addition, it is difficult to obtain intact single cells from some samples (e.g., brain tissue), so it is necessary to extract a single nucleus for sequencing. At present, many sequencing technologies [16][17][18][19]have been optimized for single-nucleus sequencing.
Single-cell isolation is a crucial point in single-cell sequencing. In the early days, the common method to isolate single cells is plate-based isolation. Single cells are picked manually with microcapillary pipettes under the microscope or sorted by flow cytometry, and then distributed into individual wells of a 96/386-well capture plate, and prepared for the subsequent operations [20]. Manual cell picking is time-consuming, low-throughput, inefficient, and requires a certain degree of micromanipulation skill. Fluorescence-activated cell sorting (FACS), a widely used technique for cell sorting, can process thousands of cells in a short time, which greatly increases the throughput, but it is hard to operate for non-professionals. In addition, >10,000 cells are required as a starting input [21]; therefore, FACS should be used conservatively when analyzing rare samples. Currently, microfluidic-based single-cell manipulation methods have become the mainstream methods of cell isolation, including droplet-based methods, microwell-based methods and the commercialized Fluidigm C1 integrated fluidic circuit (IFC) system.
Due to the limited amount of nucleic acid in a single cell, RNA molecules need to be amplified after RT to meet the requirements of sequencing. The general methods of amplification include exponential amplification based on polymerase chain reaction (PCR), and linear amplification based on in vitro transcription (IVT). PCR-based amplification can easily amplify a large number of cDNAs in a short time. However, since PCR is an exponential process that creates amplification bias, the excessive amplification of some sequences, and insufficient amplification of others, will result in inaccurate transcript quantification, and amplification errors will be propagated permanently if not properly corrected [22][23]. Linear amplification is thought to be more accurate and reproducible than PCR [24]. The second method, IVT, was first introduced by Eberwine [25]. In this strategy, an upstream T7 promoter sequence, which is used to initiate IVT, is included in the RT primer. After RT and second-strand cDNA synthesis, T7 polymerase recognizes the T7 promoter and catalyzes transcription to produce more RNA molecules. Amplified RNAs need to be reconverted into DNAs for sequencing library construction. Therefore, this method is time-consuming (~28 h starting from IVT), and increases the complexity of operation, so it is not as widely used as PCR [1].
In addition to the technical difficulties in library construction, the development of analysis pipelines that are valid for scRNA-seq data analysis is also an important technical challenge. Summaries of sequencing data analysis can be obtained in other reviews [26][27][28], and are not covered here.
2. Developmental Course of scRNA-seq
2.1. Scaling of Sequencing Throughput
Since Tang et al. performed RNA sequencing in single mouse blastomeres, for the first time, in 2009 [35], many complex questions about living systems composed of cells have been answered by scRNA-seq [20][29][30]. One of the main focal points is how many types of cells are present in functional tissues. Identifying all kinds of cell types in an unbiased and accurate manner, especially those with a low proportion, requires the researchers to analyze a large number of cells [31]. With the efforts of many researchers, scRNA-seq technology has developed from low-throughput mode to high-throughput mode. Figure 2 shows several landmark techniques that mark the development of scRNA-seq technology in terms of throughput.
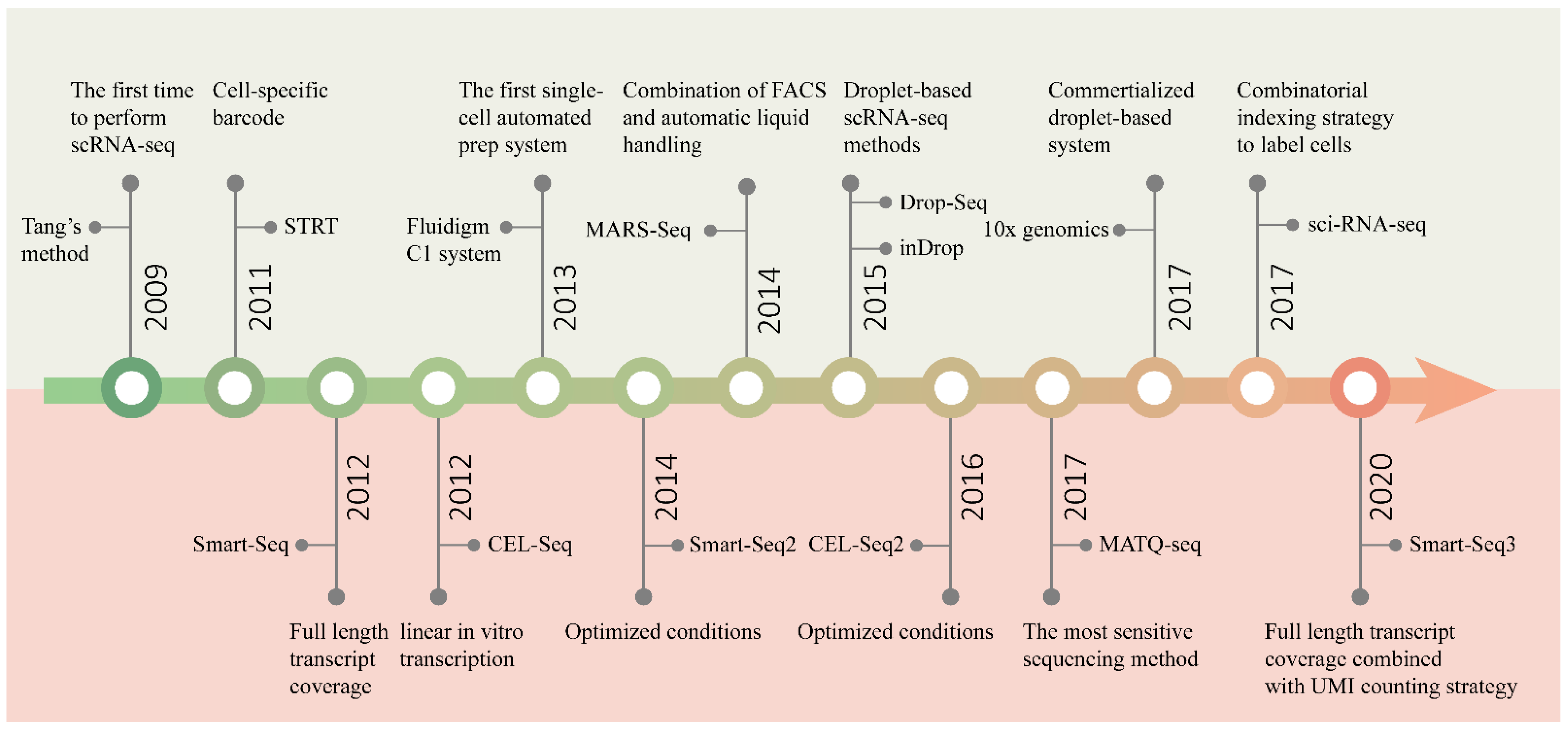
Figure 2. Timeline of the development of scRNA-seq technology. The upper half of the graph shows the main events marking the development of sequencing throughput, and the bottom half the improvement in sensitivity.
The initial scRNA-seq methods can only analyze a few cells at a time, because of the time-consuming and laborious manual cell isolation and separate sequencing library construction. In order to pool all of the transcripts from different cells for library construction and sequencing, while preserving information about their cellular origin, protocols such as STRT-seq [32] and CEL-Seq [1] introduced cell-specific barcodes. All transcripts from a single cell are labeled with the same barcode, which is unique for each cell. The barcode is a short oligonucleotide sequence that can be identified through sequencing. According to the barcode information, transcripts can be easily assigned to the corresponding cells. Barcoding is an excellent strategy to achieve parallel processing. However, the introduction of barcodes alone cannot solve the difficulty of isolating a large number of cells. Later, by combining FACS with automatic liquid handling, MARS-Seq [33] successfully sequenced thousands of cells at one time. Three levels of barcodes are used to tag mRNAs, cells and plates, respectively, to pool all the materials for the subsequent automated processing. Similarly, STRT-Seq-2i [34] utilizes a specialized FACS and barcoding protocol to increase the scale of sequencing. A custom aluminum plate with 9600 wells arranged in 96 subarrays is constructed for two rounds of barcode addition, allowing 9600 cells to be sequenced in parallel. However, these plate-based methods are not easy to carry out, and the number of cells analyzed is still limited.
With the introduction of microfluidic technology, the technical barriers of high-throughput single-cell operations have been fundamentally solved. The first commercial automated microfluidic platform, the Fluidigm C1 system, enables 96 single cells to be automatically processed at one time [35]. Nonetheless, its processing capacity is far from meeting the needs of large-scale analysis. In 2015, the emergence of two droplet-based scRNA-seq technologies [36][2] was a revolutionary breakthrough in the single-cell sequencing field, enabling the simultaneous processing of thousands of cells, and truly realizing high-thrughput parallel sequencing. Based on microfluidic droplets, the commercial platform 10x Genomics Chromium [37] was developed rapidly, which can characterize tens of thousands of cells at a time. The three technologies will be described in detail below.
More recently, sci-RNA-seq [38] and SPLiT-seq [39] using a combinatorial indexing strategy to label cells were successively published. Instead of physical compartmentation of single cells, the purpose of labeling more than 100,000 single-cell transcriptomes at one time can be achieved simply by multiple rounds of splitting and pooling. These methods are very easy to operate, have high cell labeling efficiencies, and can considerably reduce the cost of sequencing. Furthermore, the more cells sequenced at one time, the lower the cost of sequencing per cell.
2.2. Improvement in Sensitivity
In addition to the capacity to sequence multiple cells in parallel, sensitivity, accuracy, repeatability, technical noise, cost and other features are also important aspects to be considered in the design of scRNA-seq methods. Especially, sensitivity is the most critical feature, being a fundamental indicator of the performance of a method. The main events marking the development of sensitivity are shown in Figure 2.
Sensitivity can be interpreted as the probability of capturing a particular transcript and eventually detecting it by sequencing [40]. For some transcripts with low expression levels, drop-out events occur frequently in low-sensitivity sequencing platforms [41]. Low sensitivity will also reduce the accuracy and repeatability of transcript quantification, which is detrimental for the distinction of subtle differences between cell subpopulations and accurate cell type classification [42]. Every step of sequencing library construction may cause the loss of transcripts, thereby impairing the sensitivity of sequencing methods. The primary aim of all methods is to convert mRNA into cDNA for amplification. Most methods, including Smart-Seq [43] and CEL-Seq [1], use poly(T) primers to capture mRNA through the poly(A) tail to initiate RT reactions. While it can selectively capture mRNA and easily filter out numerous rRNA molecules, it also excludes some important transcripts without a poly(A) tail, such as circRNA, miRNA and nascent RNA. By contrast, MATQ-seq [44] uses random primers to capture transcripts, which can not only detect all types of transcripts, but also improve the mRNA capture efficiency, and thus achieve higher sensitivity. Another whole-transcriptome analysis method, SUPeR-seq [45], also takes advantage of random primers. After first-strand cDNA synthesis, in those methods that use PCR for cDNA amplification, the addition of the second PCR handle is also a key step in determining the efficiency of conversion. Some methods use a transferase to add a homopolymer tail, such as the poly(A) tail in SUPeR-seq or the poly(C) tail in MATQ-seq, to the 3′ end of the first-strand cDNA. A poly(T) or poly(G) primer containing the second PCR handle anneals to the homopolymer tail for second-strand synthesis. Smart-Seq uses a more convenient approach known as template-switching to incorporate another PCR handle. This method can obtain the full-length transcripts and reduce the 3′-end bias that exists in homopolymer tailing approach. However, the efficiencies of both these reactions are not 100%, and a proportion of cDNAs will inevitably be lost. To address this problem, Seq-Well S3 [46] uses random primers to initiate second-strand cDNA synthesis, and recovers most cDNA molecules without the second PCR handle. In addition, avoiding the loss of nucleic acid molecules during operation contributes substantially to the improvement in sensitivity. This is particularly evident in high-throughput methods. Cell fixation is necessary in some sequencing methods, resulting in the loss of transcripts and impaired sensitivity. For this reason, methods based on combinatorial indexing, such as sci-RNA-seq and SPLiT-seq, cannot completely replace the microfluidic-based method, despite having many benefits.
The microfluidics method reduces the reaction volume from microliters to nanoliters; thus, the sensitivity will be improved, along with an increased concentration of the targets, i.e., transcriptomes from single cells. However, when optimized, the sensitivities of the reactions in the tubes can reach the same levels as those using microfluidics. The application of microfluidics in scRNA-seq is key to improving the throughput. To improve the sensitivity, the researchers still need to focus on the fundamental chemistry utilized in scRNA-seq methods. The key to improving the sensitivity of scRNA-seq can be split into two aspects: (1) increasing the capture efficiency of RNA during the first-strand synthesis; (2) increasing the efficiency of the conversion of cDNA into amplifiable products, regardless of using second-strand synthesis or the template-switching activity of RT enzymes. Therefore, by integrating scRNA-seq chemistry with microfluidics, a higher sensitivity could be more easily achieved; meanwhile, the target concentration is greatly increased and background signal is reduced. Other smart strategies improving accuracy, reducing cost and so on will be discussed in the introduction of specific methods.
References
- Tamar Hashimshony; Florian Wagner; Noa Sher; Itai Yanai; CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Reports 2012, 2, 666-673, 10.1016/j.celrep.2012.08.003.
- Evan Z. Macosko; Anindita Basu; Rahul Satija; James Nemesh; Karthik Shekhar; Melissa Goldman; Itay Tirosh; Allison R. Bialas; Nolan Kamitaki; Emily M. Martersteck; et al.John J. TrombettaDavid A. WeitzJoshua R. SanesAlex K. ShalekAviv RegevSteven A. McCarroll Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202-1214, 10.1016/j.cell.2015.05.002.
- Yohei Sasagawa; Itoshi Nikaido; Tetsutaro Hayashi; Hiroki Danno; Kenichiro D Uno; Takeshi Imai; Hiroki R Ueda; Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biology 2013, 14, R31-R31, 10.1186/gb-2013-14-4-r31.
- Fuchou Tang; Catalin Barbacioru; Yangzhou Wang; Ellen Nordman; Clarence Lee; Nanlan Xu; Xiaohui Wang; John Bodeau; Brian B Tuch; Asim Siddiqui; et al.Kaiqin LaoAzim Surani mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods 2009, 6, 377-382, 10.1038/nmeth.1315.
- Kinga Matuła; Francesca Rivello; Wilhelm T. S. Huck; Single‐Cell Analysis Using Droplet Microfluidics. Advanced Biosystems 2019, 4, e1900188, 10.1002/adbi.201900188.
- S Curran; J A McKay; H L McLeod; G I Murray; Laser capture microscopy. Molecular Pathology 2000, 53, 64-68, 10.1136/mp.53.2.64.
- Michael R. Emmert-Buck; Robert F. Bonner; Paul D. Smith; Rodrigo F. Chuaqui; ZhengPing Zhuang; Seth R. Goldstein; Rhonda A. Weiss; Lance A. Liotta; Laser Capture Microdissection. Science 1996, 274, 998-1001, 10.1126/science.274.5289.998.
- Vibhav Gautam; Sourav Chatterjee; Ananda K. Sarkar; Single Cell Type Specific RNA Isolation and Gene Expression Analysis in Rice Using Laser Capture Microdissection (LCM)-Based Method. null 2021, 2238, 275-283, 10.1007/978-1-0716-1068-8_18.
- Kathryne Melissa Keays; Gregory P. Owens; Alanna M. Ritchie; Donald H. Gilden; Mark P. Burgoon; Laser capture microdissection and single-cell RT-PCR without RNA purification. Journal of Immunological Methods 2005, 302, 90-98, 10.1016/j.jim.2005.04.018.
- Susanne Nichterwitz; Geng Chen; Julio Aguila Benitez; Marlene Yilmaz; Helena Storvall; Ming Cao; Rickard Sandberg; Qiaolin Deng; Eva Hedlund; Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nature Communications 2016, 7, 12139, 10.1038/ncomms12139.
- Fredrik Pontén; Cecilia Williams; Gao Ling; Afshin Ahmadian; Monica Nistér; Joakim Lundeberg; Jan Pontén; Mathias Uhlén; Genomic analysis of single cells from human basal cell cancer using laser-assisted capture microscopy. Mutation Research/Mutation Research Genomics 1997, 382, 45-55, 10.1016/s1383-5726(97)00008-3.
- Jun Chen; Shengbao Suo; Patrick Pl Tam; Shengbao Suo Jing-Dong J Han; Guangdun Peng; Jun Chen Guangdun Peng Naihe Jing; Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nature Protocols 2017, 12, 566-580, 10.1038/nprot.2017.003.
- Lim Chee Liew; Yan Wang; Marta Peirats Llobet; Oliver Berkowitz; James Whelan; Mathew G. Lewsey; Laser-Capture Microdissection RNA-Sequencing for Spatial and Temporal Tissue-Specific Gene Expression Analysis in Plants. Journal of Visualized Experiments 2020, 162, e61517, 10.3791/61517.
- Sabrina Zechel; Pawel Zajac; Peter Lönnerberg; Carlos F Ibáñez; Sten Linnarsson; Topographical transcriptome mapping of the mouse medial ganglionic eminence by spatially resolved RNA-seq. Genome Biology 2014, 15, 486-486, 10.1186/s13059-014-0486-z.
- Xianwen Ren; Guojie Zhong; Qiming Zhang; Lei Zhang; Yujie Sun; Zemin Zhang; Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Research 2020, 30, 763-778, 10.1038/s41422-020-0353-2.
- Naomi Habib; Inbal Avraham-Davidi; Anindita Basu; Tyler Burks; Karthik Shekhar; Matan Hofree; Sourav R Choudhury; François Aguet; Ellen Gelfand; Kristin Ardlie; et al.David A WeitzOrit Rozenblatt-RosenFeng ZhangAviv Regev Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods 2017, 14, 955-958, 10.1038/nmeth.4407.
- Naomi Habib; Yinqing Li; Matthias Heidenreich; Lukasz Swiech; Inbal Avraham-Davidi; John J. Trombetta; Cynthia Hession; Feng Zhang; Aviv Regev; Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 2016, 353, 925-928, 10.1126/science.aad7038.
- Benjamin Lacar; Sara B. Linker; Baptiste N. Jaeger; Suguna Rani Krishnaswami; Jerika Barron; Martijn J. E. Kelder; Sarah L. Parylak; Apuã C. M. Paquola; Pratap Venepally; Mark Novotny; et al.Carolyn O'ConnorConor FitzpatrickJennifer ErwinJonathan Y. HsuDavid HusbandMichael J. McConnellRoger LaskenFred H. Gage Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nature Communications 2016, 7, 11022, 10.1038/ncomms11022.
- Kelvin See; Wilson L. W. Tan; Eng How Lim; Zenia Tiang; Li Ting Lee; Peter Y. Q. Li; Tuan D. A. Luu; Matthew Ackers-Johnson; Roger S. Foo; Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nature Communications 2017, 8, 1-13, 10.1038/s41467-017-00319-8.
- David T. Paik; Sangkyun Cho; Lei Tian; Howard Y. Chang; Joseph C. Wu; Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nature Reviews Cardiology 2020, 17, 457-473, 10.1038/s41569-020-0359-y.
- Byungjin Hwang; Ji Hyun Lee; Duhee Bang; Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine 2018, 50, 1-14, 10.1038/s12276-018-0071-8.
- Eguzkine Ochoa; Verena Zuber; Nora Fernandez-Jimenez; Jose Ramon Bilbao; Graeme R Clark; Eamonn R Maher; Leonardo Bottolo; MethylCal: Bayesian calibration of methylation levels. Nucleic Acids Research 2019, 47, e81-e81, 10.1093/nar/gkz325.
- Daniel Aird; Michael G Ross; Wei-Sheng Chen; Maxwell Danielsson; Timothy Fennell; Carsten Russ; David B Jaffe; Chad Nusbaum; Andreas Gnirke; Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biology 2011, 12, R18-R18, 10.1186/gb-2011-12-2-r18.
- Chongyi Chen; Dong Xing; Longzhi Tan; Heng Li; Guangyu Zhou; Lei Huang; X. Sunney Xie; Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189-194, 10.1126/science.aak9787.
- Jacqueline Morris; Jennifer M. Singh; James H. Eberwine; Transcriptome Analysis of Single Cells. Journal of Visualized Experiments 2011, 50, e2634, 10.3791/2634.
- Michael S. Balzer; Ziyuan Ma; Jianfu Zhou; Amin Abedini; Katalin Susztak; How to Get Started with Single Cell RNA Sequencing Data Analysis. Journal of the American Society of Nephrology 2021, 32, 1279-1292, 10.1681/asn.2020121742.
- Dominic Grün; Alexander van Oudenaarden; Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799-810, 10.1016/j.cell.2015.10.039.
- Yan Wu; Kun Zhang; Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nature Reviews Nephrology 2020, 16, 408-421, 10.1038/s41581-020-0262-0.
- Efthymia Papalexi; Rahul Satija; Single-cell RNA sequencing to explore immune cell heterogeneity. Nature Reviews Immunology 2017, 18, 35-45, 10.1038/nri.2017.76.
- Richard K. Perez; M. Grace Gordon; Meena Subramaniam; Min Cheol Kim; George C. Hartoularos; Sasha Targ; Yang Sun; Anton Ogorodnikov; Raymund Bueno; Andrew Lu; et al.Mike ThompsonNadav RappoportAndrew DahlCristina M. LanataMehrdad MatloubianLenka MaliskovaSerena S. KwekTony LiMichal SlyperJulia WaldmanDanielle DionneOrit Rozenblatt-RosenLawrence FongMaria Dall’EraBrunilda BalliuAviv RegevJinoos YazdanyLindsey A. CriswellNoah ZaitlenChun Jimmie Ye Single-cell RNA-seq reveals cell type–specific molecular and genetic associations to lupus. Science 2022, 376, eabf1970, 10.1126/science.abf1970.
- Daniel L Stoler; Carleton C Stewart; Paul C Stomper; Breast epithelium procurement from stereotactic core biopsy washings: flow cytometry-sorted cell count analysis.. Clinical Cancer Research 2002, 8, 428–432.
- Saiful Islam; Una Kjällquist; Annalena Moliner; Pawel Zajac; Jian-Bing Fan; Peter Lönnerberg; Sten Linnarsson; Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Research 2011, 21, 1160-1167, 10.1101/gr.110882.110.
- Diego Adhemar Jaitin; Ephraim Kenigsberg; Hadas Keren-Shaul; Naama Elefant; Franziska Paul; Irina Zaretsky; Alexander Mildner; Nadav Cohen; Steffen Jung; Amos Tanay; et al.Ido Amit Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776-779, 10.1126/science.1247651.
- Hannah Hochgerner; Peter Lönnerberg; Rebecca Hodge; Jaromir Mikes; Abeer Heskol; Hermann Hubschle; Philip Lin; Simone Picelli; Gioele La Manno; Michael Ratz; et al.Jude DunneSyed HusainEd LeinMaithreyan SrinivasanAmit ZeiselSten Linnarsson STRT-seq-2i: dual-index 5ʹ single cell and nucleus RNA-seq on an addressable microwell array. Scientific Reports 2017, 7, 1-8, 10.1038/s41598-017-16546-4.
- Daniel M. DeLaughter; The Use of the Fluidigm C1 for RNA Expression Analyses of Single Cells. Current Protocols in Molecular Biology 2018, 122, e55-e55, 10.1002/cpmb.55.
- Allon M. Klein; Linas Mazutis; Ilke Akartuna; Naren Tallapragada; Adrian Veres; Victor Li; Leonid Peshkin; David A. Weitz; Marc W. Kirschner; Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187-1201, 10.1016/j.cell.2015.04.044.
- Grace X. Y. Zheng; Jessica M. Terry; Phillip Belgrader; Paul Ryvkin; Zachary W. Bent; Ryan Wilson; Solongo B. Ziraldo; Tobias D. Wheeler; Geoff P. McDermott; Junjie Zhu; et al.Mark T. GregoryJoe ShugaLuz MontesclarosJason G. UnderwoodDonald A. MasquelierStefanie Y. NishimuraMichael Schnall-LevinPaul W. WyattChristopher M. HindsonRajiv BharadwajAlexander WongKevin D. NessLan W. BeppuH. Joachim DeegChristopher McFarlandKeith R. LoebWilliam J. ValenteNolan G. EricsonEmily A. StevensJerald P. RadichTarjei S. MikkelsenBenjamin J. HindsonJason H. Bielas Massively parallel digital transcriptional profiling of single cells. Nature Communications 2017, 8, 14049, 10.1038/ncomms14049.
- Junyue Cao; Jonathan S. Packer; Vijay Ramani; Darren A. Cusanovich; Chau Huynh; Riza Daza; Xiaojie Qiu; Choli Lee; Scott N. Furlan; Frank J. Steemers; et al.Andrew AdeyRobert H. WaterstonCole TrapnellJay Shendure Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661-667, 10.1126/science.aam8940.
- Alexander B. Rosenberg; Charles M. Roco; Richard A. Muscat; Anna Kuchina; Paul Sample; Zizhen Yao; Lucas T. Graybuck; David J. Peeler; Sumit Mukherjee; Wei Chen; et al.Suzie H. PunDrew L. SellersBosiljka TasicGeorg Seelig Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176-182, 10.1126/science.aam8999.
- Christoph Ziegenhain; Beate Vieth; Swati Parekh; Björn Reinius; Amy Guillaumet-Adkins; Martha Smets; Heinrich Leonhardt; Holger Heyn; Ines Hellmann; Wolfgang Enard; et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell 2017, 65, 631-643.e4, 10.1016/j.molcel.2017.01.023.
- Peter V. Kharchenko; Lev Silberstein; David T. Scadden; Bayesian approach to single-cell differential expression analysis. Nature Methods 2014, 11, 740-742, 10.1038/nmeth.2967.
- Christian H. Holland; Jovan Tanevski; Javier Perales-Patón; Jan Gleixner; Manu P. Kumar; Elisabetta Mereu; Brian A. Joughin; Oliver Stegle; Douglas A. Lauffenburger; Holger Heyn; et al.Bence SzalaiJulio Saez-Rodriguez Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biology 2020, 21, 1-19, 10.1186/s13059-020-1949-z.
- Daniel Ramsköld; Shujun Luo; Yu-Chieh Wang; Robin Li; Qiaolin Deng; Omid R Faridani; Gregory A Daniels; Irina Khrebtukova; Jeanne F Loring; Louise C Laurent; et al.Gary P SchrothRickard Sandberg Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology 2012, 30, 777-782, 10.1038/nbt.2282.
- Kuanwei Sheng; Wenjian Cao; Yichi Niu; Qing Deng; Chenghang Zong; Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nature Methods 2017, 14, 267-270, 10.1038/nmeth.4145.
- Xiaoying Fan; Xiannian Zhang; Xinglong Wu; Hongshan Guo; Yuqiong Hu; Fuchou Tang; Yanyi Huang; Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biology 2015, 16, 1-17, 10.1186/s13059-015-0706-1.
- Travis K. Hughes; Marc H. Wadsworth; Todd M. Gierahn; Tran Do; David Weiss; Priscila R. Andrade; Feiyang Ma; Bruno J. De Andrade Silva; Shuai Shao; Lam C. Tsoi; et al.Jose Ordovas-MontanesJohann E. GudjonssonRobert L. ModlinJ. Christopher LoveAlex K. Shalek Second-Strand Synthesis-Based Massively Parallel scRNA-Seq Reveals Cellular States and Molecular Features of Human Inflammatory Skin Pathologies. Immunity 2020, 53, 878-894.e7, 10.1016/j.immuni.2020.09.015.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
5.5K
Revisions:
3 times
(View History)
Update Date:
01 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No